

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Traumatic brain injury (TBI) is a critical public health problem worldwide and one of the most important causes underlying high mortality due to traffic accidents, falls, and external mechanical concussion etc. [18,30]. Typically, TBI is defined as an alteration of brain function and/or evidence of brain pathology, caused by an external force [18]. It has long been recognized that TBI can cause significant motor and cognitive impairments, that are common, persistent, and costly. Because of the morbidity and mortality, the molecular pathways which trigger pathophysiological processes after TBI remain to be explained clearly [4]. TBI has two distinct phases : acute, primary injury and delayed, secondary injury. Primary injury occurs at the trauma instant and comprises brain damage and diffuse axonal injury. Secondary injury occurs hours to days or months after the initial trauma and includes mitochondrial dysfunction, lipid peroxidation, ionic imbalance, excessive release of neurotransmitters, and membrane degradation, all of which contribute to neuronal cell loss. Thus, the discovery of novel agents could serve the purpose and provide beneficial effect against TBI [27].
Recent studies have suggested that nicotinamide mononucleotide adenylyl transferase 2 (NMNAT2), one of three naturally occurring mammalian NMNATs, is highly expressed in brain, heart, liver, and skeletal muscle of SD rats [19]. Besides, NMNAT2 is a sensitive enzyme marker for nicotinamide adenine dinucleotide levels that reflects the intracellular redox equilibrium and cellular energy state, acting as a sensor for cells with a high energy demand, including cancer cells [13,20]. But whether NMNAT2 have neuroprotective effect in TBI is still unconfirmed. Since neuronal apoptosis plays an essential role following TBI, and NMNAT2 is demonstrated as a protective component in a number of neurodegenerative disorders such as Parkinson’s disease, motor neuron disease and Alzheimer’s disease, we hypothesized that NMNAT2 might also be neuroprotective against secondary brain damage after TBI [11].
In this study, we investigated whether expression of NMNAT2 was changed in injured-side cortex after TBI induced by a weight-dropping method in a rat model. We also assessed if NMNAT2 was protective against TBI-induced neuronal apoptosis.
MATERIALS AND METHODS
Animals
All animal experiments were approved by the Institutional Animal Care Committee of Soochow University (ZJGYYLL-2020-04-003) and were implemented according to the guidelines of the National Institutes of Health on the care and use of laboratory animals.
Eighty-seven male Sprague-Dawley (SD) rats (280–310 g) were purchased from the Animal Center of Chinese Academy of Sciences, Shanghai, China. The animals were housed in a temperature- and humidity-controlled animal environment with food and water available ad libitum on a 12-hour light-dark cycle.
Experimental rat TBI model
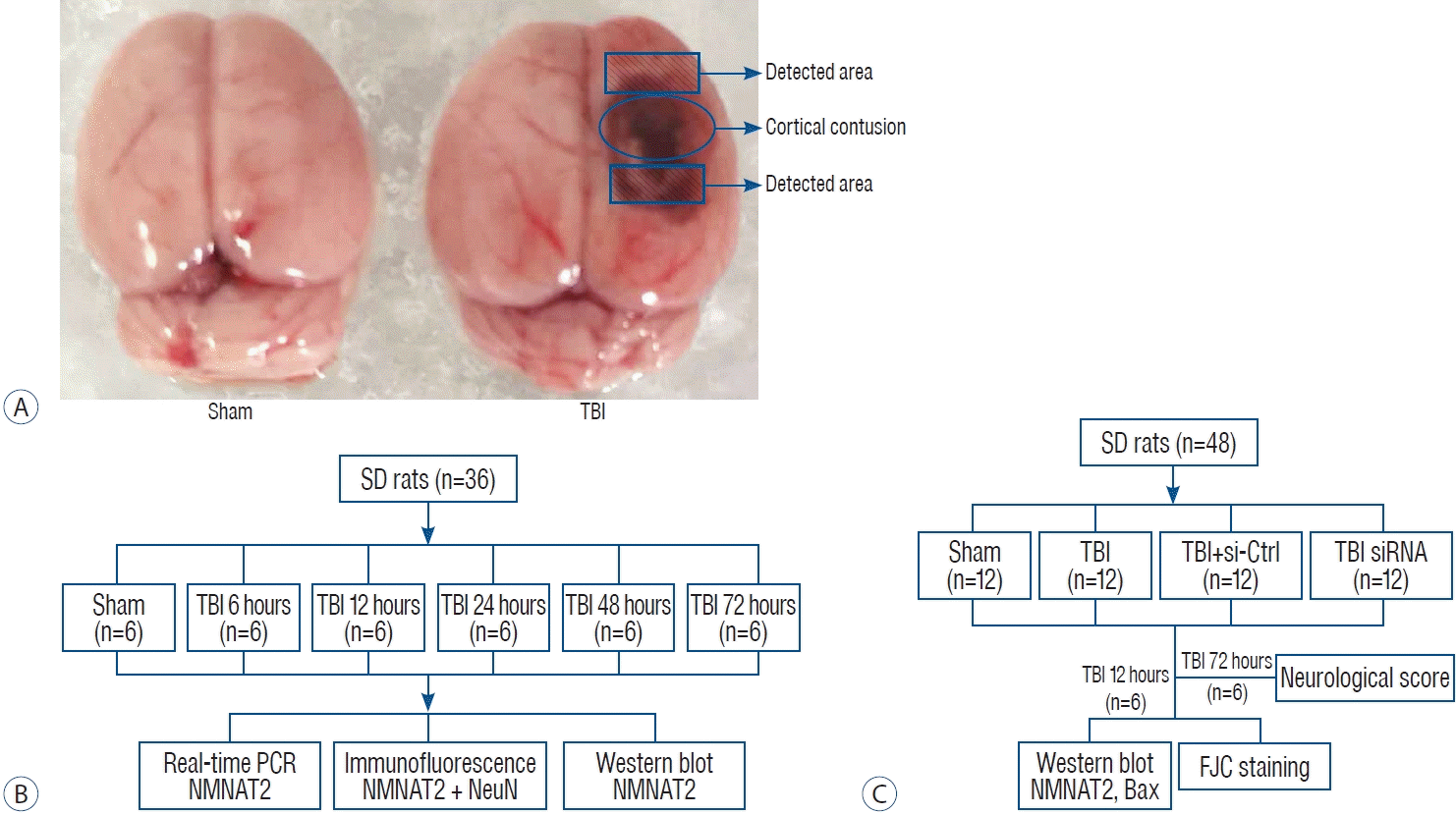
The rat TBI model was established using a weight-drop method as described previously [16]. Animals were anesthetized with isof lurane inhalation (3% induction, 1–2% maintenance). Following successful anesthesia, the animal’s head was fixed on a stereotactic apparatus. Immediately behind incision of the scalp, the skin and fascia were retracted, and a 5 mm diameter craniotomy was drilled 2.5 mm next to right side of the midline and 1.5 mm after the right coronal suture. The bone flap was removed and the dura mater was kept intact. A controlled cortical severe impact injury was made by dropping a 40 g weight cylindrical steel rod onto a piston (3 mm diameter tip, 2.8 mm depth) resting on the exposed dura from a height of 25 cm. The impact force was 1000 g/cm, inducing an injury belonged to severe TBI. Then the skin was sutured and the animals were taken back to their home cage after recovery in a warm room. The sham groups underwent identical initial procedures, including craniotomy, but without the weight drop.
Experimental design
Experiment I
To analyze the relative expression of NMNAT2 at different time points after TBI. Male SD rats (38 rats were modeled but only 36 rats survived surgery) were randomly distributed into a sham-treated control group, and groups sacrificed at 6, 12, 24, 48, and 72 hours post-TBI (n=6 each). Rats were sacrificed at the pre-set time points, and the tissue samples surrounding the contusion cortex were collected for subsequent analysis such as real-time polymerase chain reaction (PCR), western blot and immunofluorescence staining (Fig. 1B).
Experiment II
To investigate the role of NMNAT2 in TBI induced-brain damage, 48 rats (excluding one that did not survive the surgery) were randomly distributed into four groups (sham, TBI, TBI + small-interfering [siRNA]-control, TBI + siRNA-NMNAT2; n=12 for each group). Based on the results of the experiment I, rats were sacrificed 12 hours after TBI, and the tissue samples surrounding the contusion cortex were obtained for western blot, and Fluoro-Jade C (FJC) staining. Additionally, we conducted neurological score testing 72 hours after TBI (Fig. 1C).
Tissue collection and sectioning
The animals were anesthetized with 3% isoflurane inhalation at an identical time point after TBI and then transcardially perfused with a 4°C solution of 0.9% saline. Protein and RNA were extracted from cortex samples surrounding the injured-area (Fig. 1A).
After transcardial perfusion with a 4°C solution of 0.9% saline, the animals were transcardially perfused with 250 mL of ice-cold 4% paraformaldehyde (pH 7.4). Brain tissues were quickly fixed in 4% paraformaldehyde for 24 hours, then sequentially dehydrated in phosphate-buffered saline (PBS) with 15% and 30% sucrose. Frozen coronal sections near the contusion area were collected using a freezing microtome at a thickness of 15 µm (Leica Microsystems, Wetzlar, Germany).
Transfection
In vivo siRNA transfection in the rat brain was performed according to the manufacturer’s instructions for Entranster in vivo RNA interference transfection reagent (Engreen, 18668- 11-2; Engreen Biosystem Co. Ltd., Beijing, China). Specific NMNAT2-siRNA and Control-siRNA were obtained from Guangzhou Ribo Biotechnology Co. Ltd. (Guangzhou, China). Briefly, 500 pmol (6 µL) of NMNAT2-siRNA and controlsiRNA were respectively dissolved in 13 µL of transfection reagent. Intracerebroventricular injection was administered as previously described [24]. Twenty-four hours before TBI, rats were anesthetized with isoflurane inhalation (3% induction, 1–2% maintenance) and placed in a stereotaxic apparatus. Then we injected 19 µL mixture (0.5 µL/min) by using a 50- µL Hamilton syringe (Hamilton Company, Reno, NV, USA) through a burr hole into the right lateral ventricles which was located at 1.5 mm posterior, 1.0 mm lateral to the bregma, and 3.2 mm below the horizontal plane of the skull.
Real-time PCR
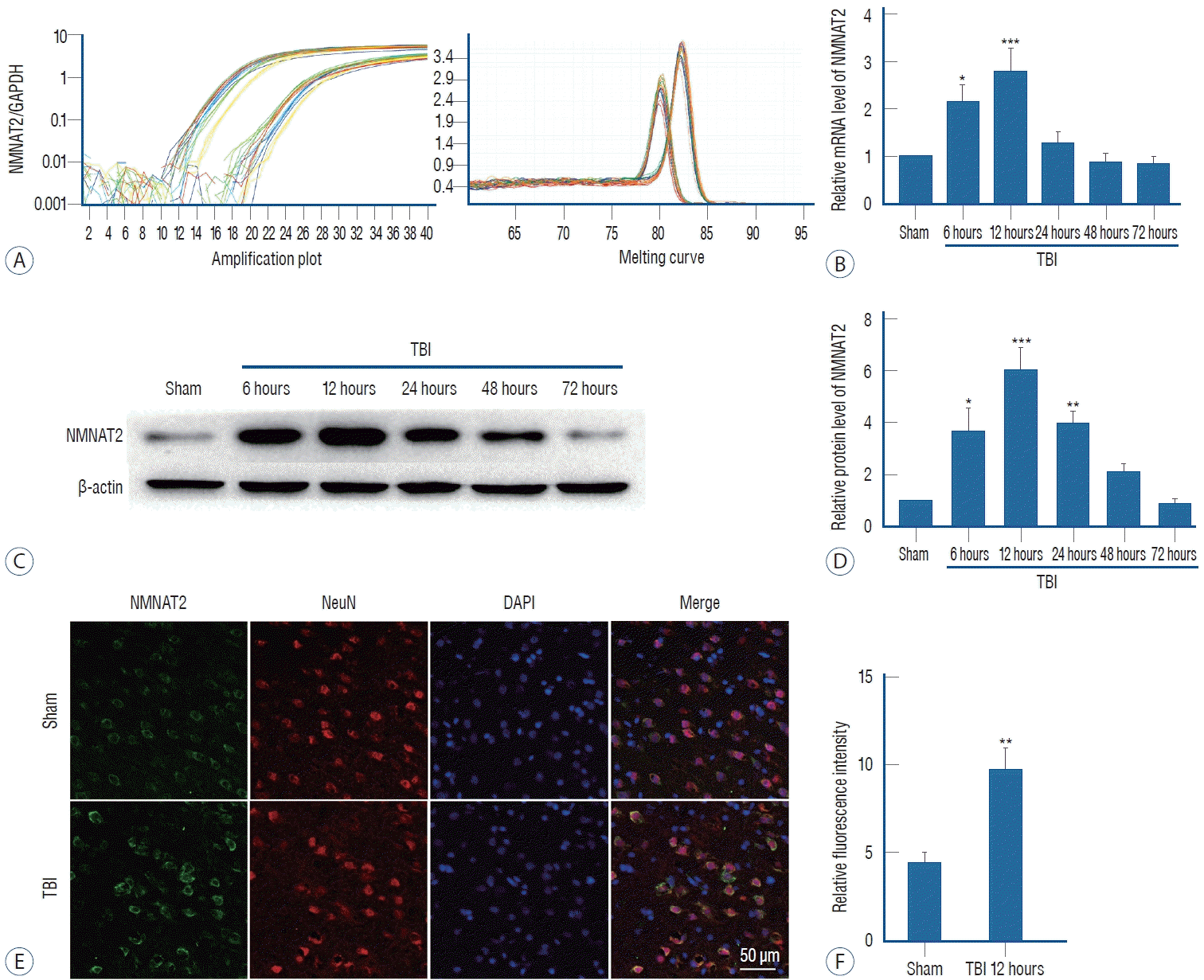
Total RNA was extracted from contusion-side cortex using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. About 1 µg total RNA was reversely transcribed into cDNA by using a cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. In real-time PCR, a PowerUpTM SYBRTM Green Master Mix kit (Thermo Fisher Scientific) was used to amplify cDNA, and the fluorescent signal intensity of SYBR Green quantitatively represented the amount of double-stranded DNA present. The primer sequences of NMNAT2 and GAPDH are as follows : NMNAT2, L : 5’-GCATCCTCTCCAACGGTCAAC-3’, R : 5’-TTCTGGTAAATGGGCTGGGT-3’; GAPDH, L : 5’-TGGCCTTCCGTGTTCCTACC-3’, R : 5’-CGCCTGCTTCACCACCTTCT-3’.
Western blotting
For total protein extraction, the contusion-side cortex of animals was lysed completely in ice-cold radioimmunoprecipitation lysis buffer (Beyotime, Shanghai, China) that contained protease inhibitor with centrifugation at 12000 g at 4°C for 20 minutes. The supernatant was collected, and then protein concentration was estimated using a bicinchoninic acid assay kit (23227; Thermo Fisher Scientific). After denaturing at 95°C for 5 minutes in 5× loading sodium dodecyl sulfate (SDS) buffer (Invitrogen), a total of 30 µg protein of each sample was separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA, USA). Blocking buffer containing 5% non-fat milk was used to block the membranes for 1 hour at room temperature. Then the membranes were incubated with primary antibodies against β-actin (1 : 10000), NMNAT2 (Abcam, Cambridge, UK; 1 : 1000), BCL-2-associated X protein (Bax; 1 : 500; Protein Tech, Rosemont, IL, USA) at 4°C overnight. The next day, the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase. Finally, the immunoblots were detected by enhanced chemiluminescence reagents (Millipore) in an imaging system (BioRad, Hercules, CA, USA). The relative protein intensity was analyzed using Image-J software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence staining
In sham and 12 hours post-TBI groups, double immunofluorescence staining for NMNAT2 and NeuN was performed to evaluate neuronal expression of NMNAT2. After three washes (10 minutes each) in PBS with 0.3% Triton X-100 (Biofroxx, Essen, Germany), the brain sections were blocked with 10% goat serum for 1 hour at room temperature, followed by incubation at 4°C overnight with primary antibodies : rabbit anti-NMNAT2 (1 : 200; Abcam), and mouse anti-NeuN (MAB377, 1 : 300; Millipore). The following day, the sections were washed three times with PBS, then were incubated with fluorescent secondary antibodies (1 : 800 Alexa Fluor 488 donkey anti-rabbit immunoglobulin [Ig] G antibody, and Alexa Fluor 555 donkey anti-mouse IgG antibody; Invitrogen) for 1 hour at room temperature. After washing three times with PBS, sections were covered with 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI). Finally we detected immunofluorescence staining using a laser confocal microscope Leica DMi8 (Leica Microsystems).
FJC staining
Neuronal degeneration in brain sections were detected by FJC staining according to the manufacturer’s instructions (Millipore). After treatment with 1% sodium hydroxide in 80% alcohol for 5 minutes, the sections were incubated in 70% alcohol for 2 minutes and rinsed with double distilled water (ddH2O) for 2 minutes. Then the sections were transferred to 0.06% potassium permanganate for 10 minutes and rinsed with ddH2O for 2 minutes. Next, the sections were immersed in FJC working solution (0.1% acetic acid) for 20 minutes followed by rinsed in ddH2O water twice and dried in an oven (50°C) for 5–8 minutes. Subsequently, sections were washed in xylene for at least 1 minute before they were covered with resin. Finally, the sections were inspected with a laser confocal microscope (Leica DMi8; Leica Microsystems).
Statistical analysis
All data were presented as mean±standard error of mean and analyzed using SPSS ver. 19.0 software (IBM Corp., Chicago, IL, USA). One-way analysis of variance combined with Tukey’s multiple-comparisons test and Student’s t test were used to analysis all results. p<0.05 was considered to statistically significant.
RESULTS
NMNAT2 expression was significantly increased in injured-side cortex after TBI
To assess NMNAT2 expression levels, we performed realtime PCR and western blot in the sham group and for groups sacrificed at 6, 12, 24, 48, and 72 hours after TBI. We found that NMNAT2 mRNA expression increased significantly at 6 hours and reached the peak 12 hours after TBI. Then expression decreased quickly and was close to the level of the sham group at 48 hours after TBI (Fig. 2A and B). The protein expression of NMNAT2, mirrored that of mRNA, it was increased at 6 hours and peaked 12 hours after TBI, then decreased gradually. However, the protein level, declined slowly and had a baseline similar to sham group until 72 hours after TBI (Fig. 2C and D). Based on the results of real-time PCR and western blot, we performed immunofluorescence staining in samples collected 12 hours after TBI. NMNAT2 fluorescence intensity of NMNAT2 was also increased 12 hours post-TBI, which was consistent with the western blot results (Fig. 2E and F).
NMNAT2 inhibition aggravated TBI induced neuronal degeneration
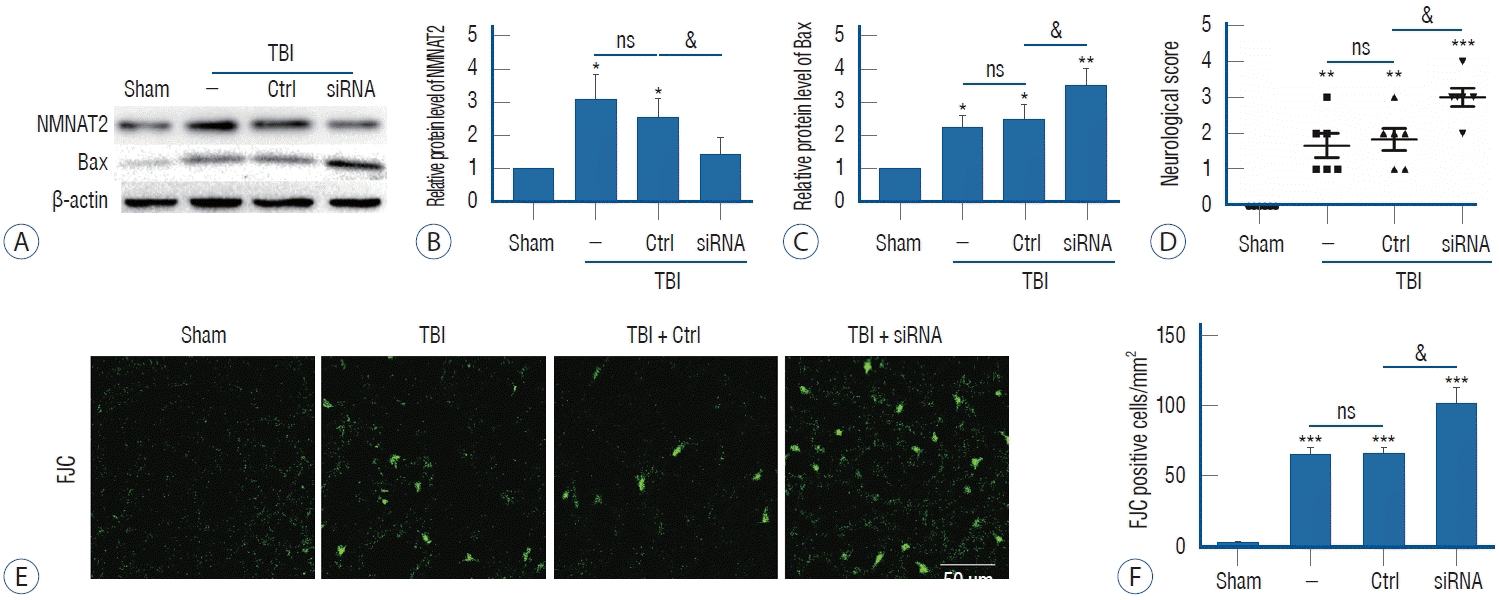
The effect of intraventricular transfection with NMNAT2 siRNA was confirmed by western blot. Our results revealed that intraventricular transfection with NMNAT2 siRNA effectively reduced NMNAT2 protein expression 12 hours after TBI (Fig. 3A and B). To investigate the anti-neurodegenerative effect of NMNAT2 after TBI, experiment II evaluated the protein level of Bax, which had been reported to promote neurodegeneration after TBI. We found that Bax protein level was significantly upregulated after TBI, and it was further overexpressed when the TBI model was treated with NMNAT2 siRNA (Fig. 3A and C). FJC staining was also performed to explore the role of NMNAT2 in TBI-induced neuronal degeneration. TBI was associated with a higher number of FJC-positive cells in the injured-side cortex, and NMNAT2 siRNA remarkably increased the number of FJC-positive cells after TBI (Fig. 3E and F). These data suggest that NMNAT2 inhibition can aggravate TBI-induced neuronal degeneration and implicates activation of the Bax signaling pathway.
Inhibition of NMNAT2 aggravated neurological deficits
Neurological behavior is widely impaired following TBI. In this study, we attempted to verify whether inhibition of NMNAT2 effected neurological behavior after TBI. Compared with the TBI group and control siRNA group, when NMNAT2 was down-regulated, neurological function scores were significantly higher (Fig. 3D). This suggested that inhibition of NMNAT2 could aggravate neurological deficits after TBI.
DISCUSSION
TBI is the devastating pathological condition that mainly occurs because of mechanical injury [12]. Pathophysiological responses of secondary brain injury, including inflammation, oxidative damage, apoptosis, structural axonal injury, and brain edema, are the potential therapeutic targets of TBI [14]. In the present study, we confirmed that NMNAT2 expression was increased and NMNAT2 exhibited neuroprotective activity via attenuating neuronal apoptosis and degeneration after TBI.
Whether NMNAT2, a gatekeeper of axonal degeneration, could be neuroprotective in TBI-induced secondary injury is the main reason why we explored this study. NMNAT is the biosynthetic enzyme of NAD+, which is a catalytic enzyme for adenosine-triphosphate synthesis. Each of the three mammalian NMNAT isoforms, NMNAT1, NMNAT2, and NMNAT3, delay neuronal degeneration when overexpressed [15,21]. Up to now, the three NMNAT isoforms show differences in subcellular localization and tissue distribution [3], but little is known about the effects of the NMNAT family members on brain injury after TBI. Among the three NMNAT isoforms, NMNAT2 is reported to be most sensitive to NAD and can act as a sensor to intracellular NAD level [15], which might partly explain the facts that high levels of NMNAT2 are detected in the organs with high demand for energy such as brain, heart, and skeletal muscle [3]. NMNAT2 is synthesized in the Golgi, then transported down the axons. It has a short half-life and can be quickly degraded in neuron after injury [17]. In vitro studies revealed that activated mitogen-activated protein kinase signaling post-injury could lead to the loss of NMNAT2 and activation of sterile alpha and TIR motif containing 1 (SARM1), and subsequently NAD+was depleted prior to neuronal degeneration [6,29]. There are neuroprotective roles for several NAD+ related metabolites [9]. Recent studies demonstrated that NAD+loss is the consequence of SARM1 activation [8,25]. In vivo studies showed that knock out of SARM1 can improve functional outcomes after traumatic injury in mice [10]. Our results revealed that the mRNA and protein levels of NMNAT2, significantly increased in the injuredside cortex after TBI and peaked 12 hours after TBI. NMNAT2 up-regulation in the injured-side area appeared to be the early damage response to TBI. NMNAT2 mRNA expression then obviously decreased at 24 hours after TBI. However, protein expression declined much more slowly. This might be due to the decreased ubiquitination and degradation of NMNAT2 after TBI [2,28].
To explore the role of NMNAT2 post-TBI, we administered NMNAT2 siRNA via intracerebroventricular injection 24 hours before TBI. Notably, NMNAT2 inhibition was associated with more neurodegeneration (FJC staining) at 12 hours post-TBI and worse neurological functional outcome at 72 hours post-TBI. At the molecular level, the protein level of Bax in the TBI + NMNAT2 siRNA group was remarkably higher than in the TBI + control siRNA group at 12 hours post-TBI. These results indicate that NMNAT2 can ameliorate neurodegeneration after TBI and the Bcl-2/Bax signaling pathway is involved. Bax is a pro-apoptotic member of the Bcl-2 family and is normally located in cytoplasm. In response to proapoptotic stimuli, Bax translocate to the outer mitochondrial membrane to increase mitochondrial outer membrane permeability. This leads to release of cytochrome C from the mitochondrial intermembrane to the cytoplasm. Next, cytochrome C binds with caspase-activating molecule (Apaf-1), inducing the sequential activation of the caspase cascade including caspase 3 and 9, which results in apoptosis [1,7]. As a key activator of apoptosis, Bax plays a critical role in neuronal apoptosis after TBI [5,22,23]. These findings are consistent with our results that NMNAT2 inhibition can aggravate TBI induced neuronal degeneration and implicates activation of the Bax signaling pathway.
This study is somewhat limited. Though the results in our study that the NMNAT2 siRNA group had worse outcome are encouraging, they do not clarify the exact mechanism by which NMNAT2 prevent TBI induced neuronal apoptosis. Thus, further studies may need to be performed to help understanding the mechanism. Moreover, there are no direct evidence that the poor outcome in NMNAT2 siRNA group was the result of more neuronal degeneration, and functional assays that directly determine the contribution of the functional integrity to neurological deficits will be required in our further studies.
CONCLUSION
Our study demonstrates that NMNAT2 mRNA and protein levels dramatically increased on injured-side cortex 12 hours post-TBI. Inhibition of NMNAT2 enhances neurodegeneration and neurological deficits post-TBI, and this process involves activation of Bax signaling. Based on these findings, we propose that NMNAT2 might be a therapeutic target for secondary brain injury following TBI.
 XML Download
XML Download