

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Remifentanil, which has a rapid onset time and fast clearance, is extensively utilized for intraoperative analgesia [1,2]. However, remifentanil may elicit remifentanil-induced hyperalgesia (RIH) [3-5]. Although extensively studied, the mechanisms responsible for RIH remain largely unknown.
N-methyl-d-aspartate receptors (NMDARs) arecrucialinmodulating excitatory synaptic transmissions. RIH is associated with NMDAR-mediated central sensitization [6–8]. However, the pathway underlying the spinal NR2B subunit of NMDAR upregulation after remifentanil infusion remains unknown.
Excessive production of reactive oxygen species (ROS) has detrimental effects on cell metabolic and other physiological activities [9]. Dynamin-related protein 1 (DRP1) induces excess mitochondrial fission, resulting in a temporal redox imbalance and the overproduction of ROS [10]. Several studies have found that ROS are involved in neuropathic pain via spinal mechanisms [11,12]. ROS have also been reported to participate in opioid tolerance coupled with hyperalgesia [13,14].
Owing to their high energy demands, neurons exhibit physiological characteristics that depend on mitochondrial function [15]. DRP1 is a cytosolic GTPase that migrates between the cytosol and mitochondrial network to dock to the mitochondrial outer membrane [16]. DRP1 forms ring-like structures and mediates mitochondrial fission [17]. A mitochondrial-derived ROS outburst was recently documented to lead to postoperative RIH by regulating NR2B subunit phosphorylation in the spinal cord [18]. However, whether DRP1 contributes to RIH via the DRP1-mitochondria-ROS pathway is still unknown.
The authors hypothesized that the spinal inhibition of the DRP1-mitochondria-ROS-NMDA pathway had the effect of reversing the outburst of NR2B subunit expression in the dorsal horn, thereby providing a theoretical basis for RIH treatment.
MATERIALS AND METHODS
1. Animals
Experiments were conducted on ninety adult male Sprague–Dawley rats (180–230 g). All rats were acquired from the Department of Experimental Animal Sciences and acclimated for at least seven days with an adequate supply of food and water. Animals were housed six per cage, in an air-conditional animal room (23°C ± 2°C) with a 12/12 hour light/dark cycle. Behavioraltests were performed between 8:00 A.M. and 12:00 P.M. The experiments were approved by the Animal Care Committee of Wenzhou Medical University (No. WYDW2019-0549) and adhered to the guide for the care and use of laboratory animals. The experiments were also performed in compliance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines 2.0. The animals were deeply anesthetized using sevoflurane before they were euthanized.
2. Intrathecal catheter placement
Under sevoflurane anesthesia (HengRui Co.) via a nose mask, the back was disinfected with 10% povidone-iodine solution. At the L3-4 level, a longitudinal incision (1 cm) was made along the midline. The tissue was separated with forceps until the spinous process was exposed. Without spinous process removal, ophthalmic forceps were used to detect the access to the intrathecal space, which was confirmed by the tail-flickreflex of the rat. Then, a sterilized PE-10 tube was placed along the access between the L3-4 interlaminar space and situated at the lumbar enlargement. After hemostasis, the incision was disinfected with iodine and sutured [19]. After one week of recovery, experiments were performed. The rats that did not show hind limb paralysis after intrathecal administration of lidocaine were excluded from the study [20].
3. Establishment of the RIH model
Under sevoflurane anesthesia, remifentanil was infused at a rate of 1.2 μg/kg/min for 60 minutes via the tail vein [20,21]. Meanwhile, a longitudinal incision was made from the proximal edge of the heel extending toward the toes in the right hind paw. The plantaris muscle was elevated using forceps, leaving the muscle origin intact. After hemostasis, the incision was disinfected and sutured [22].
4. Test drugs
Remifentanil (Humanwell Pharma Co.) was diluted to 10 μg/mL before use. The antisense oligodeoxynucleotides (ODNs) against DRP1 (AS-DRP1) mRNA, 5’CCA CTA CGA CAA TCT GAG GC3’, was designed to knock down dorsal DRP1 expression. The mismatch ODNs to DRP1 (MM-DRP1) mRNA was 5’ ACA CTT CTA CAT TCG GAC GC3’. MM-DRP1 had six mismatched bases compared with AS-DRP1 [23]. These ODNs were obtained from the Shenggong Company.
AS-DRP1 and MM-DRP1 were dissolved in nuclease-free water at 2 μg/μL and kept at –80°C. Thirty minutes before remifentanil infusion, they were administered at 40 μg, followed by 10 μL nuclease-free water flush through a PE-10 tube (Smith Medical, Inc.) [23].
5. Behavior testing
Paw withdrawal mechanical thresholds (PWMT) were evaluated using von Frey filaments (Stoelting Co.). Rats were placed in plastic cages (10 × 20 × 9 cm) and separated by thin partitions, with a wire mesh bottom (0.7 × 0.7 cm). The von Frey filaments were used to apply different bending forces (1, 1.4, 2, 4, 6, 8, 10, 15, and 26 g) to the plantar surface of the hind paw. Paw licking or withdrawal was considered as nociceptive-like response. In order to turn the ordinal data into continuous data, the PWMT was determined by the “up-and-down” method [24–26]. The pain threshold was calculated according to the formula: PMWT = . Xf = log value of the last test, the k value was obtained from the k-value table, δ = 0.231.
A radiant heat source (Stoelting Co.) delivered a thermal stimulus to the wound to evaluate the paw withdrawal thermal latency (PWTL). The PWTL was defined as the time to withdraw the hind paw. To avoid unexpected injury, a maximum cutoff time of 25 seconds was used [21].
Behavioral tests were repeated three times at 5-minute intervals between each test. The mean PWMT and PWTL were obtained by averaging these values. Each rat was acclimatized for 30 minutes before testing. The behavioral operator was blinded to the groups of rats.
6. Western blotting
Rats were euthanized before collecting the right side of their L4-6 spinal cord. The samples were isolated and kept in liquid nitrogen. Tissue samples were sonicated on ice in lysis buffer containing protease inhibitors (Sigma–Aldrich Co.). The lysate was centrifuged at 4°C (11,000 rpm, 25 min), and the supernatant was pipetted as the total protein. Samples were separated by 8% or 12% sulfonate–polyacrylamide gel electrophoresis. According to the loading marker, the gels containing DRP1, GAPDH, and NR2B proteins were cut and transferred to polyvinylidene difluoride membranes (Millipore). After blocking with nonfat milk (5%) for 1.5 hours on a shaker, the blots were incubated overnight at 4°C with the following rabbit primary antibodies: anti-DRP1 (1:1,000, 8570, CST), anti-GAPDH (1:1,000, 2083, CST), and anti-NMDAR2B (1:1,000, 4212, CST). After three washes with TBST, the membrane was incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:10,000, BI003A, Biosharp) at 25°C for 60 minutes [20]. The density of the specific bands was calculated by a Chemi-Dos XRS imaging system (FluorChem FC3; ProteinSimple) and quantified using AlphaView software (ProteinSimple).
7. Immunofluorescence technique
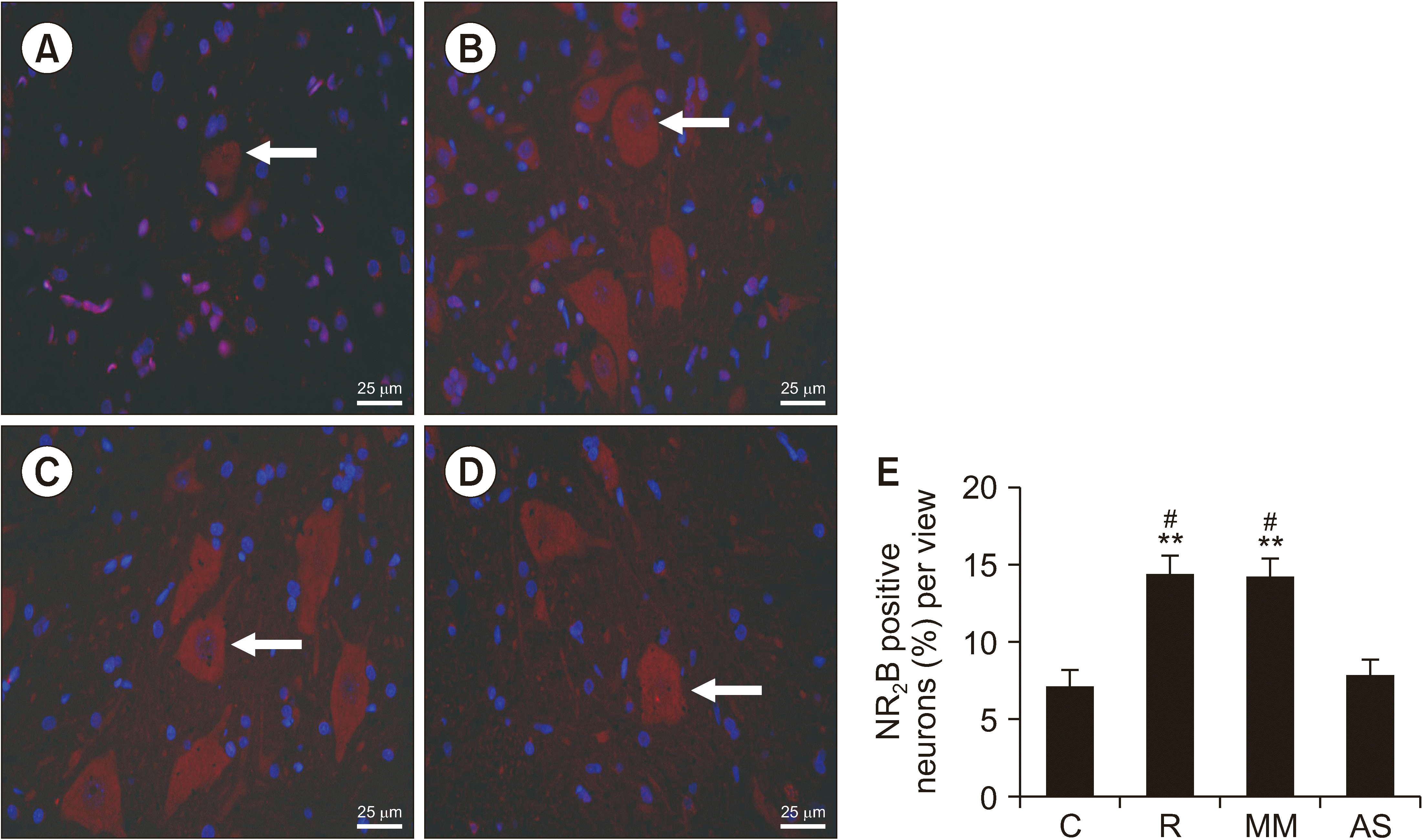
Under sevoflurane anesthesia, twelve rats were perfused, and the right side of the spinal L4-6 segments were removed, followed by postfixation for two hours in 4% paraformaldehyde (PFA), and maintained in 30% sucrose for 48 hours at 4°C. The spinal L4-6 segments were sectioned at 25 µm and stored in 0.01 M phosphate buffered saline (PBS). The spinal cord samples were blocked for 1 hour in 5% donkey serum at room temperature. The sections were then incubated overnight with anti-NR2B (1:50, 4212, Cell Signaling Technology) at 4°C. After washing with PBS, the goat anti-rabbit secondary antibody (Proteintech; 1:100) was used to incubate for 2 hours at room temperature. After washing with PBS, the sections were mounted on glass slides using Fluoromount-G (Fisher Scientific) and imaged with a confocal microscope [18].
8. Mitochondrial superoxide imaging
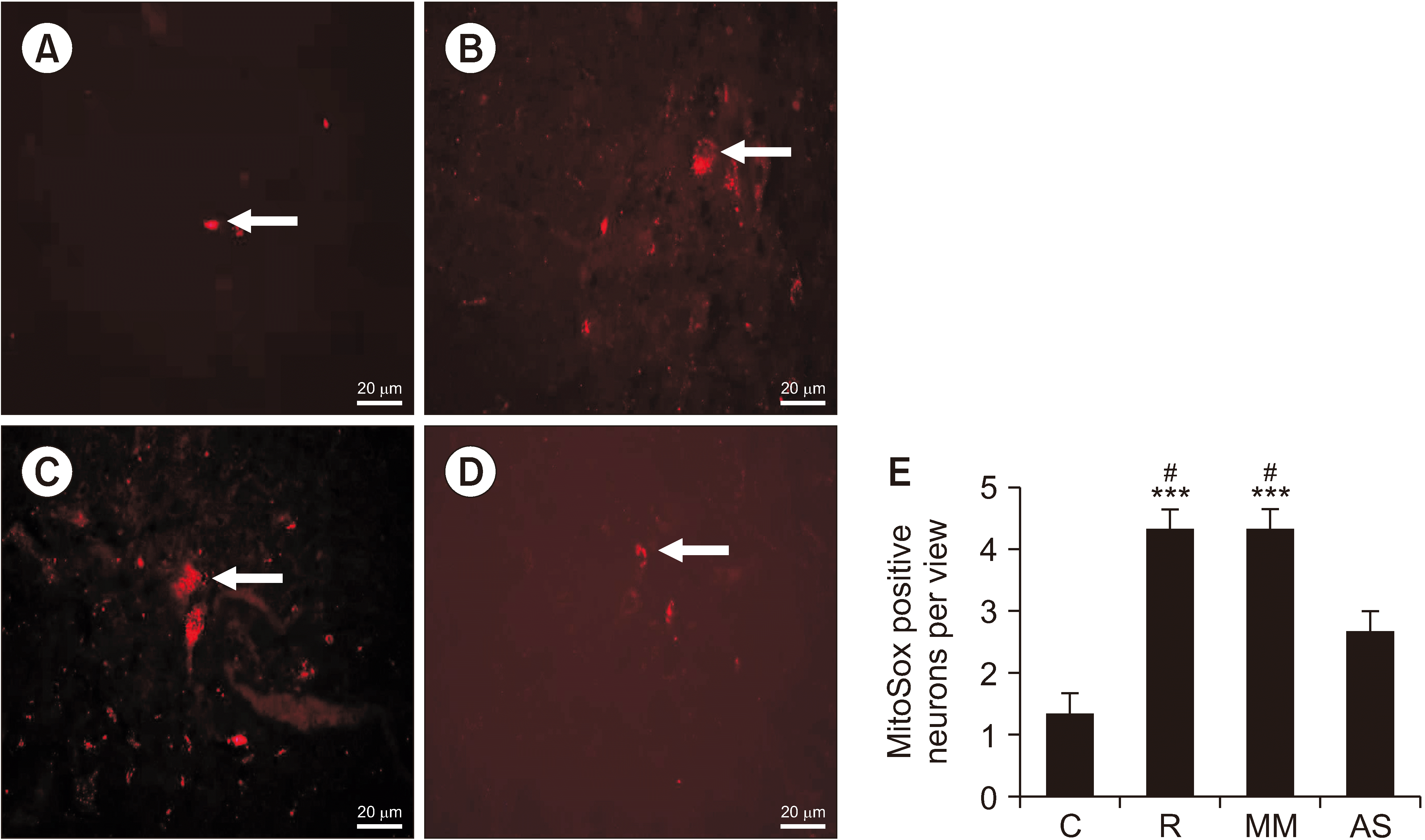
An indicator of mitochondrial superoxide (MitoSox red; Invitrogen) was dispersed in a 1:1 mixture of dimethyl sulfoxide with 0.9% NaCl (33 μM in 30 μL) [27]. Seventy minutes after MitoSox intrathecal injection, rats were intracardially injected with 4% PFA. After perfusion, the right side of the spinal L4-6 segments were removed, followed by postfixation overnight in 4% PFA and cryoprotection with 30% sucrose in PBS for the next two days. They were then sectioned at 35 μm and examined using fluorescence microscopy with a rhodamine filter. The number of MitoSox-positive cells was quantified as described previously [27,28].
9. Electron microscopy
Under sevoflurane anesthesia, the rats were perfused via the right ventricle using normal saline. The right side of the spinal L4-6 segments were removed and chopped into blocks on ice (< 1 mm3). They were then fixed at 4°C for 3 hours in 2.5% glutaraldehyde. Subsequently, timely treatment of the specimens was ensured to prevent cell autolysis by various internal enzymes. Thereafter, the samples were dehydrated in 70%, 80%, 90%, and 100% acetone, followed by osmic acid treatment and embedding on a porous template. A teacher in the electron microscope room helped make specimen pieces into copper. The number, structure, and morphology of the mitochondria were observed using transmission electron microscopy. After the right field of vision was selected, photographs were used to record and quantify the mitochondria (Media Cybernetics, Inc.) [29].
10. Experimental protocol
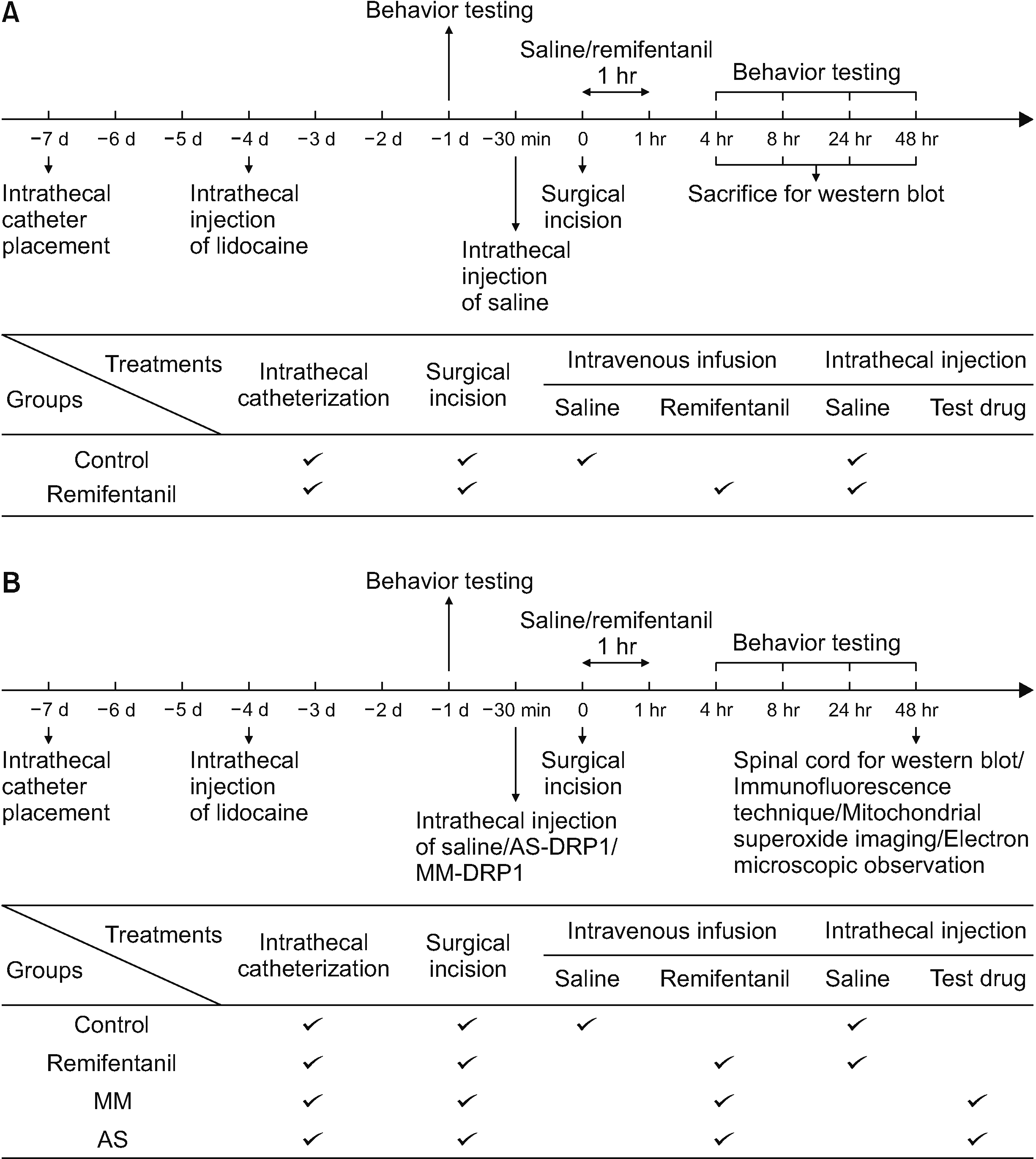
1) Experiment 1: DRP1 expression contributed to RIH
Thirty rats were allocated to five groups by a computer random order generator (n = 6 in each group): the control group (surgical incision without remifentanil infusion), 4H group, 8H group, 24H group, and 48H group (4 hour, 8 hour, 24 hour, 48 hour after the RIH model respectively). PWTL and PWMT were evaluated at baseline (–24 hour) and 4 hour, 8 hour, 24 hour, and 48 hour after remifentanil exposure. The L4-6 spinal cord segments were obtained in 2 minutes for Western blotting after the behavioral tests. A schematic of the experiment is shown in Fig. 1A.
2) Experiment 2: The effect of AS-DRP1 on RIH
To investigate whether knockdown of DRP1 expression could prevent RIH, AS-DRP1 was used to test the effect. Sixty adult rats were randomly allocated to four groups by a computer random order generator (n = 15 in each group): C (surgical incision alone), R (remifentanil infusion and surgical incision), MM (remifentanil infusion and surgical incision plus MM-DRP1), and AS (remifentanil infusion and surgical incision plus AS-DRP1). Thirty minutes before the surgical incision, normal saline, MM-DRP1, or AS-DRP1 (20 μL) was administered by intrathecal injection.
Behavioral assessments were conducted at –24 hour and 4, 8, 24, and 48 hour after remifentanil exposure. The L4-6 segments of twenty-four rats were removed (n = 6 in four groups) for Western blotting, twelve rats (n = 3 in four groups) for immunofluorescence, twelve rats (n = 3 in four groups) for mitochondrial superoxide imaging, and twelve rats (n = 3 in four groups) for electron microscopy analyses after the last behavioral assessment. A schematic of the experiment is shown in Fig. 1B.
11. Statistical analysis
All data are presented as the mean ± standard deviation. Repeated measures analysis of variance (ANOVA) were performed to determine overall differences at each time point in PWMT and PWTL. The trapezoidal rule, that multiples the time interval with PWMT or PWTL during 48 hours after remifentanil infusion, was used to calculate the area under the curves (AUC). The AUC, Western blot and other molecular experiments were computed using one-way ANOVA. SPSS (version 23.0; IBM Co.) was used in the statistical analyses. P < 0.05 was considered statistically significant.
RESULTS
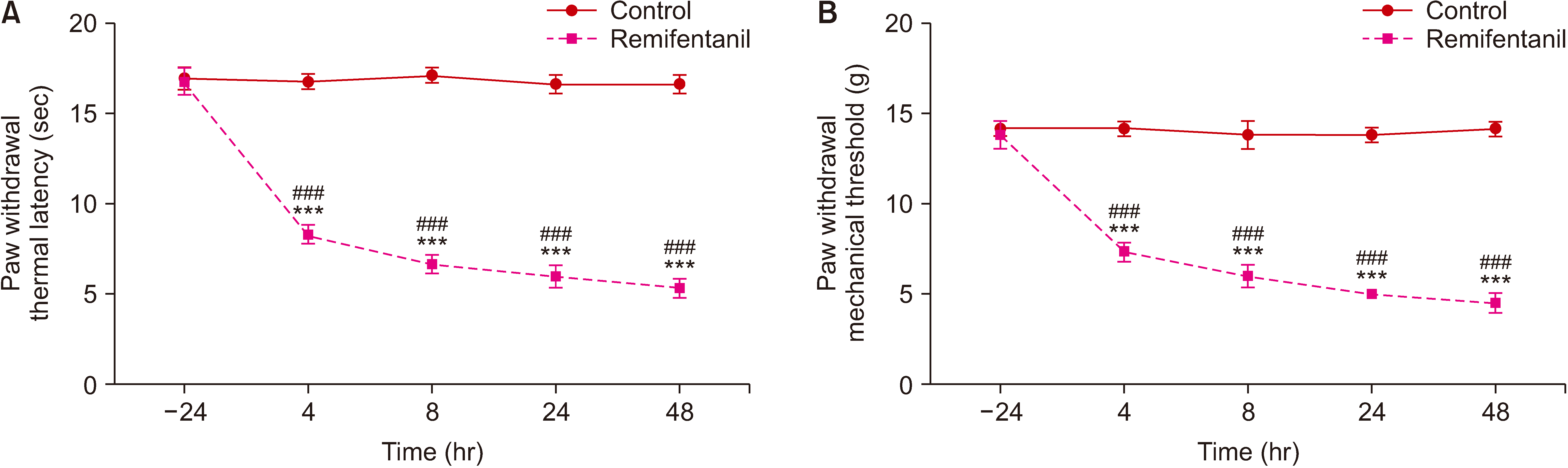
1. Remifentanil exposure decreased PWTL and PWMT values and increased NR2B and DRP1 expression
As shown in Fig. 2, before remifentanil infusion, the baseline (–24 hour) values of PWTL and PWMT were similar in the control and remifentanil groups. PWTL and PWMT in group R were decreased significantly compared with baseline from 4 to 48 hour (P < 0.001). PWTL and PWMT in group R were decreased significantly compared with group C, which started at 4 hour (PWTL in group C vs. group R: 16.83 ± 0.41 vs. 8.33 ± 0.52 second, P < 0.001 and PWMT in group C vs. group R: 14.17 ± 0.41 vs. 7.33 ± 0.52 g, P < 0.001) to 8 hour (PWTL in group C vs. group R: 17.17 ± 0.41 vs. 6.67 ± 0.52 second, P < 0.001 and PWMT in group C vs. group R: 13.83 ± 0.75 vs. 6.00 ± 0.63 g, P < 0.001), 24 hour (PWTL in group C vs. group R: 16.67 ± 0.52 vs. 6.00 ± 0.63 second, P < 0.001 and PWMT in group C vs. group R: 13.83 ± 0.41 vs. 5.00 ± 0.00 g, P < 0.001) and 48 hour (PWTL in group C vs. group R: 16.67 ± 0.52 vs. 5.33 ± 0.52 second, P < 0.001 and PWMT in group C vs. group R: 14.17 ± 0.41 vs. 4.50 ± 0.55 g, P < 0.001).
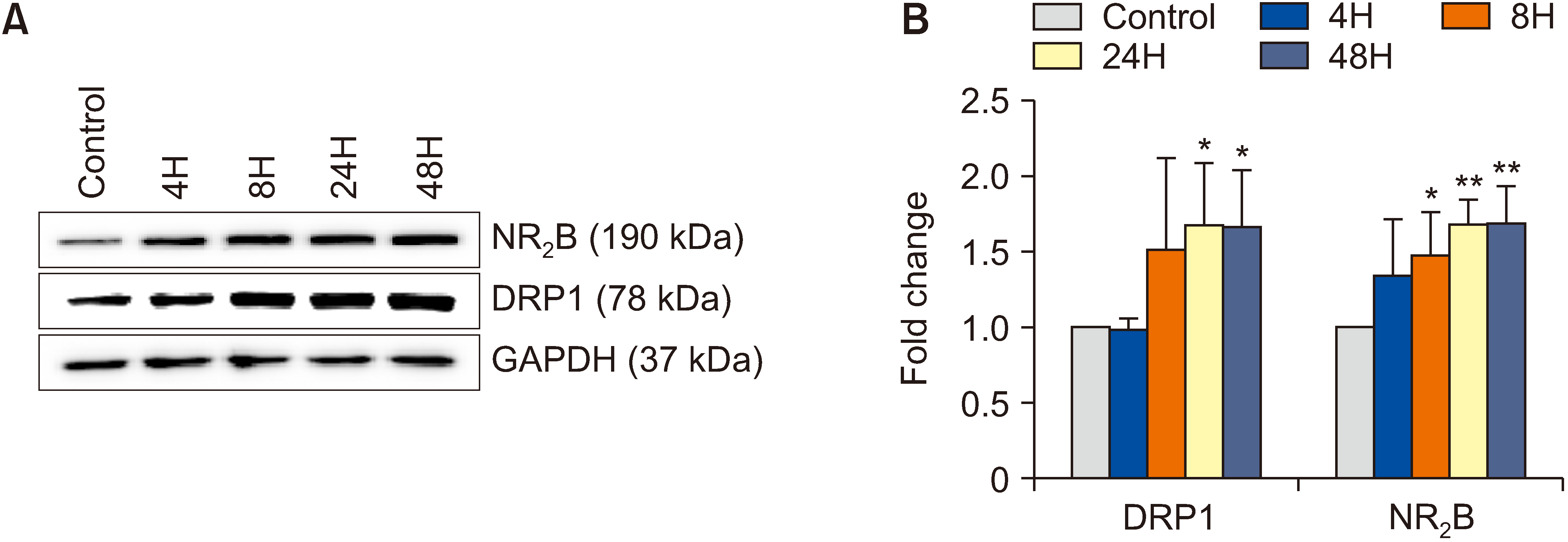
As shown in Fig. 3, Western blotting revealed time-dependent upregulation of DRP1 and NR2B expression after remifentanil exposure. Compared with that in group C, the dorsal DRP1 expression in group R was increased at 24 hour (P = 0.028) and 48 hour (P = 0.032). The expression of NR2B in group R was increased at 8 hour (P = 0.049), 24 hour (P = 0.002) and 48 hour (P = 0.005).
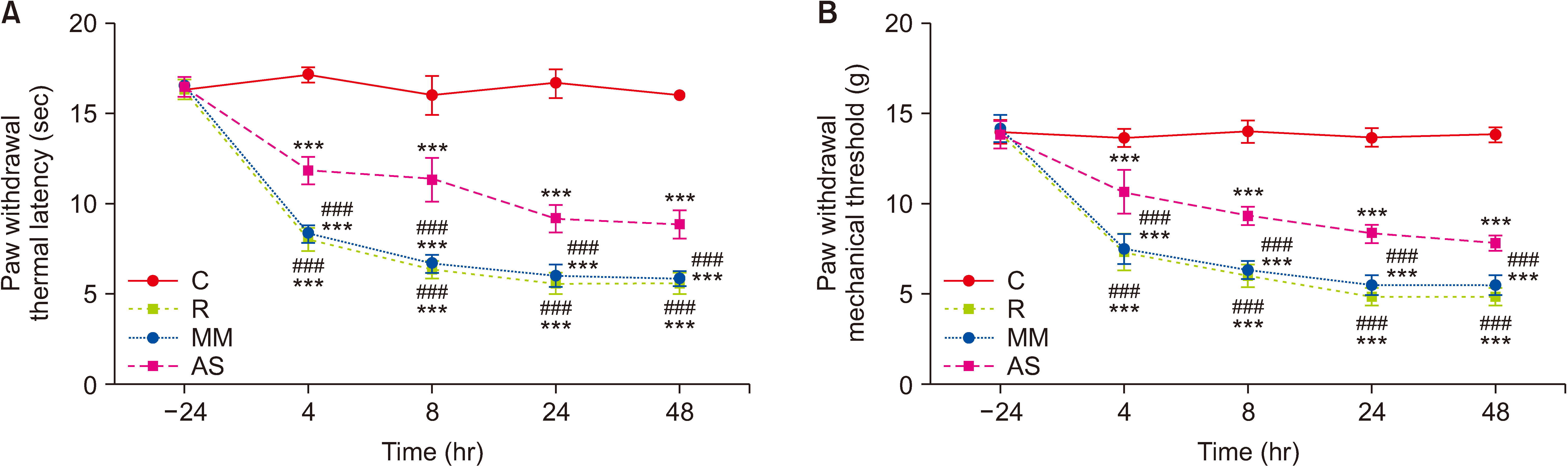
2. AS-DRP1 prevented RIH
As shown in Fig. 4, there were no significant differences in the baseline among the four groups. After 4 hour of remifentanil exposure, the PWTL and PWMT of rats (group R, group MM, and group AS) were significantly reduced compared with the control rats (P < 0.001). This finding suggested that group R, group MM, and group AS developed RIH.
As expected, PWTL and PWMT in group AS were increased from 4 hour to 48 hour compared with group R and group MM (P < 0.001, Fig. 4). Group AS had higher AUCs of PWMT and PWTL than group R and group MM (P < 0.001, Table 1). These findings indicated that pretreatment with AS-DRP1 effectively prevented remifentanil induced thermal hyperalgesia and allodynia.
3. AS-DRP1 reversed DRP1 and NR2B expression in RIH rats
After remifentanil infusion, the expression of spinal DRP1 and NR2B in group R was significantly increased compared to that in group C (P = 0.006 and P = 0.003; Fig. 5). The NR2B expression in group AS was significantly higher than that in group C (P = 0.017). Compared with group R and group MM, AS-DRP1 in group AS significantly suppressed spinal DRP1 expression (P = 0.020 in group R and P = 0.021 in group MM) and NR2B expression (P = 0.022 in group R and P = 0.013 in group MM). This finding suggested that AS-DRP1 reversed the expression of DRP1 and NR2B in RIH rats.
NR2B-positive cell numbers in group R and group MM were also significantly higher than those in group C (P = 0.009 in group R and P = 0.010 in group MM). Compared with group R and Group MM, the number of NR2B-positive cells was significantly lower in group AS (P = 0.015 in group R and P = 0.017 in group MM; Fig. 6).
4. AS-DRP1 reduced the mitochondrial number and the production of ROS
In the spinal dorsal horn, the number of mitochondria in group R was significantly higher than that in group C (P = 0.003, Fig. 7). This finding indicated that the fission of mitochondria was increased after remifentanil infusion. After intrathecal injection of MM-DRP1 and AS-DRP1, the number of mitochondria in the AS group was significantly less than that in the R and MM groups (P = 0.020 and P = 0.024, Fig. 7). It was also observed that some mitochondria in group R were at the division stage. There was no statistical difference between group MM and group R. These results suggested that AS-DRP1 effectively inhibited mitochondrial fission.
As shown in Fig. 8, the MitoSox-positive cell numbers in group R were significantly higher than those in group C (P < 0.001). This finding indicated that the ROS production was increased after the remifentanil exposure. MitoSox-positive cell numbers in group AS were significantly lower than those in group R (P = 0.031). There was no statistical difference between group MM and group R, suggesting that AS-DRP1 decreased ROS production in RIH rats.
DISCUSSION
The present research showed that 1) RIH could generate a behavioral hyperalgesic state and induce time-dependent upregulation of DRP1 and NR2B receptors in the spinal cord; 2) AS-DRP1 administration inhibited remifentanil-induced behavioral nociception and downregulated the expression of DRP1 and NR2B; and 3) AS-DRP1 suppressed mitochondrial fission and inhibited ROS outbursts. These findings support the hypothesis that AS-DRP1 modulates NMDAR expression to prevent RIH through the DRP1-mitochondria-ROS pathway.
Remifentanil is used extensively as an intraoperative analgesic. Intraoperative administration of remifentanil was correlated with increased postoperative analgesic requirements and exacerbated hyperalgesia in clinical cases [30]. However, the mechanism underlying RIH remains unclear. Excessive NMDAergic excitatory transmitters constitute neuropathic pain in various pain models [31]. It has been suggested that activation of NMDAR in the spinal dorsal horn contributes to the pathogenesis of RIH [32]. Remifentanil has been reported to directly excite NMDAR subunits in vivo [33].
The mitochondrial dynamic system, modulated by the function of DRP1, is closely associated with the mitochondrial redox status and oxidative respiratory chain. DRP1 phosphorylation under normal conditions ensures stable oxidative respiration in mitochondria. Excessive phosphorylation of DRP1 under stress conditions disrupts the oxidative balance, eventually generating excess ROS [34].
ROS, which are primarily produced by mitochondria, contribute to inflammation and oxidative stress, eventually resulting in mitochondrial damage [35]. Mitochondrial fission has been reported to mediate pathophysiological ROS production [36]. Mitochondria-derived ROS in the spinal cord are involved in the development of RIH [18].
In the current study, mitochondrial fission and ROS overproduction were detected after remifentanil exposure. The physiological concentration of ROS is maintained by a balance between ROS generation and elimination. Nonetheless, under stress conditions, DRP1 upregulation impairs ROS elimination resulting from mitochondrial fission and excessive mitochondrial fragmentation [37]. The DRP1-mitochondria-ROS cascade is linked to the initiation of neuropathic pain [38]. Therefore, it was speculated that the absence of DRP1 could lead to the downregulation of spinal NMDAR expression and hence relieve RIH.
Recent studies have correlated DRP1 downregulation with mitochondrial impairment-mediated pain. Studies have found evidence of the DRP1-mitochondria-ROS cascade during neuropathic pain. A previous study showed that the selective DRP1 repressor Mdivi-1 can abolish hydrogen peroxide-induced mechanical hyperalgesia [38]. DRP1 expression was also enhanced in a chronic constriction injury-induced pain model. Successive hyperbaric oxygen treatment can relieve hypersensitivity [29]. Antisense ODNs against DRP1 reversed the increased amount of DRP1 in the spinal cord and attenuated mechanical allodynia in a rat model of glycoprotein-induced neuropathic pain [23]. Recent studies have also shown that suppressing DRP1-mediated mitochondrial fission may relieve bone cancer pain [39,40]. However, whether DRP1 has a relationship with RIH is unknown. In the present study, as expected, remifentanil exposure was observed to upregulate the expression of DRP1 in the spinal cord. More importantly, DRP1 knockdown effectively attenuated RIH, prevented mitochondrial fission, reduced the ROS production, and downregulated the overexpression of DRP1 and NR2B. The above results indicated that DRP1 was involved in RIH. In addition, AS-DRP1 reversed RIH via the DRP1-mitochondria-ROS pathway.
However, this study also had several limitations. The findings indicated that RIH upregulated DRP1 expression, followed by abnormal mitochondrial fission. However, only protein expression alteration and mitochondrial morphological changes were tested in the trial. Further studies are required to confirm mitochondrial functional changes after remifentanil infusion.
In conclusion, the authors demonstrated that RIH is related to DRP1 protein expression. Moreover, intrathecal administration of AS-DRP1 relieved RIH-linked behavioral parameters via downregulation of spinal DRP1, inhibition of mitochondrial fission, and suppression of ROS outburst. These findings highlight the involvement of the DRP1-mitochondria-ROS-NMDA pathway in RIH pathogenesis. Spinal blockade of the DRP1 response has proven to be a potent way to prevent RIH.


 XML Download
XML Download