

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Cardiovascular diseases (CVDs) are a major health concern and the leading cause of death worldwide [1]. Despite recent advances in treatment, CVDs remain a significant burden on healthcare systems globally [2]. CVDs result in irreversible damage to the heart muscle, and current treatments have limitations in restoring damaged cardiac tissue. Therefore, there is an urgent need for new therapies that can promote cardiac regeneration [1,2]. Mesenchymal stem cells (MSCs) have gained prominence as a promising cell source for cardiac regeneration, primarily due to their abundant paracrine factors that possess angiogenic and tissue regenerative properties. However, the potential of MSCs to differentiate into cardiomyocytes, the cells responsible for heart contractions, has been reported to be limited [3-5]. MSCs can be isolated from various tissues, such as bone marrow, adipose tissue, and umbilical cord tissue, and have shown promising results in preclinical studies for cardiac regeneration [6-8].
PSME4, also known as PA200, is a protein that plays a role in regulating the activity of the proteasome, a large protein complex that degrades unwanted or damaged proteins in cells [9]. Specifically, PSME4 functions as an activator of the immunoproteasome, which is responsible for generating peptides for major histocompatibility complex (MHC) class I presentation [10]. MHC class I molecules are important for the immune system to recognize and eliminate cells that have become infected with viruses or have developed into cancer cells [11]. In addition to its role in the immune response, PSME4 has also been implicated in various cellular processes, including DNA damage response and cell proliferation [12]. It is also known that PSME4 target acetylated histone H3 for degradation, which can lead to changes in gene expression and cellular behavior in a proteasomal independent manner [13]. Besides, recent studies have shown that PSME4 plays a critical role in determining the fate of transcriptional co-activator YAP1 in the nucleus [14].
YAP1, or Yes-associated protein 1, is a transcriptional co-activator that plays a critical role in various cellular processes, including stem cell maintenance, tissue regeneration, and organ development [15,16]. YAP1 is a downstream effector of the Hippo signaling pathway, which is a conserved pathway that regulates organ size and tissue homeostasis [15,17]. YAP1 plays a critical role in stem cell biology, regulating both stem cell maintenance and differentiation [16,18-20]. YAP1 promotes the self-renewal of embryonic stem cells by promoting the expression of pluripotency factors, such as Oct4 and Nanog, and in adult stem cells, YAP1 regulates tissue homeostasis by promoting stem cell proliferation and inhibiting differentiation [20,21]. YAP1 has also been implicated in stem cell differentiation, promoting differentiation into specific cell types in certain tissues such as the heart [22]. Overall, understanding the molecular mechanisms that regulate YAP1 activity in stem cells is crucial for developing new therapies for tissue regeneration and disease treatment.
Our previous study demonstrated that apicidin, a class I histone deacetylase (HDAC) inhibitor, promotes cardiac commitment of MSCs [14,23], but the precise mechanism by which it occurs is not fully understood. We identified that the protein PSME4 recognizes and degrades acetylated-YAP1 in the nucleus during apicidin-dependent cardiac commitment. Here we propose the novel role of PSME4 in cardiac commitment by YAP1 degradation which is crucial for developing new therapies for cardiac regeneration and other diseases.
METHODS
Reagent and antibodies
Reagents used were as follows: apicidin, tubastatin A, and doxycycline were from Sigma. Puromycin was from InvivoGen. Scramble or HDAC6 siRNA were purchased from Dharmacon. Antibodies used were as follows: Anti-PSME4 was from Abcam. Anti-YAP1, anti-HDAC6, anti-acetylated-tubulin, and anti-GAPDH were from Santa Cruz Biotechnology. Anti-actin, anti-tubulin, and anti-flag were from Sigma. Alexa Fluor 568-conjugated goat anti-mouse IgG was from Molecular Probes (Invitrogen).
Genetically engineered mice
The PSME4 knockout mice utilized in this study were obtained from RIKEN Bioresource Research Center (RBRC09401) after obtaining permission from the original depositor, Professor Tomoki Chiba (University of Tsukuba) [13]. Genotyping was performed using the following oligomer set:
sense: 5ʹ-CCTCCCAAGTGTCTAAAGCCGCTTATACTG-3ʹ,
sense (neo): 5ʹ-TCGTGCTTTACGGTATCGCCGCTCCCGA TT-3ʹ,
antisense: 5ʹ-GAGACCTTCTGCACTTCCAAGGATCTCAT-3ʹ.
The wild-type mice produced 600 base pair products, while the neomycin cassette produced 1 kb bands. The animal usage for the study was reviewed and approved by the Chonnam National University Medical School Research Institutional Animal Care and Use Committee (CNU IACUC-H-2020-12).
Bone marrow MSCs
Bone marrow-derived MSCs were obtained according to previous method with slight modification. 8-week-old mice were terminated with cervical dislocation and both femurs were kept after removing adherent muscle and ligaments. The femur was prepared after cutting both ends, subsequently flushed the marrow with phosphate buffered saline (PBS) and collected the pass-through PBS. Mononuclear cells can then be isolated from the aspirates using density gradient centrifugation with Percoll and washed with serum-free media. The cells are seeded into culture dishes in Dulbecco’s modified Eagle’s medium (DMEM) high glucose with 10% fetal bovine serum and antibiotics. Incubation is carried out at 37°C in a humidified atmosphere with 5% CO2. Non-adherent cells are removed by medium change after 48 h, with subsequent changes every 2–3 days. Passaging is performed upon reaching 70%–80% confluence by detaching the cells using trypsin/EDTA.
Immortalized human MSCs
The study utilized immortalized human bone marrow-derived MSCs, which were kindly provided by Professor Yeon-Soo Kim (Inje University). The hTERT-MSCs were generated by infecting the cells with telomerase and selecting with puromycin. The cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified atmosphere of 95% air and 5% CO2. The hTERT-MSCs expressed CD90 but were negative for CD45, CD34, CD14, and CD11b. The differentiation potential of the hTERT-MSCs into adipogenic, osteogenic, or chondrogenic lineages was previously confirmed.
Immunofluorescence
Cells were seeded on cover slips and cultured in six-well plates. After being treated with specific conditions, the cells were fixed with 3.7% paraformaldehyde for 10 min and then washed with PBS. Permeabilization was achieved using 0.5% Triton X-100 in PBS for 10 min with continuous agitation. After being washed three times for 5 min with PBS, the cells were blocked for 1 h at room temperature with 1% bovine serum albumin (BSA) in PBS. Following blocking, the cells were incubated overnight with primary antibodies (YAP1, 1:1,000) in blocking buffer (1% BSA/PBS). Primary antibodies were washed with 0.1% PBS/Tween-20 three times. YAP1 antibodies were further probed with Alexa-conjugated secondary antibodies (568 for mouse). Cover slips were flipped and mounted with one drop of anti-fade solution containing 4ʹ,6-diamidino-2-phenylindole (Molecular Probes). The cells were analyzed using the NIS-Elements AT program (Nikon Inc.).
Small hairpin RNA
Lentivirus-driven small hairpin (sh) RNA was used to knock down endogenous gene expression. The desired nucleotides were cloned into either the reverse tetracycline transactivator system-controlled shuttle vector (LT3GEPIR) or a constitutively expressing shuttle vector (SGEP), and pMD2.G and psPAX2 were used to generate lentivirus. In case of LT3GEPIR, puromycin selection was further carried out. Complete culture media (10% FBS and 1% penicillin/streptomycin in DMEM high glucose) with 4 µg/ml of puromycin was changed after incubation of lentivirus overnight. Selection was terminated after two-day incubation with puromycin. Lentiviral infection and shRNA activation were monitored by detecting the expression of green fluorescent protein after either overnight incubation (in the case of SGEP) or one day of treatment with 2 µg/ml of doxycycline (in the case of LT3GEPIR). The knockdown efficiency was evaluated through Western blot or quantitative real-time polymerase chain reaction (PCR) analysis performed 2 days after infection for SGEP or one day after treatment with 2 µg/ml of doxycycline for LT3GEPIR. The nucleotide sequences utilized for the knockdown were as follows:
-
Non-targeting control:
5’-TGCTGTTGACAGTGAGCGCAGGAATTATAATGCTTATCTATAGTGAAGCCACAGATGTATAGATAAGCATTATAATTCCTATGCCTACTGCCTCGGA-3’ -
Human YAP1:
5’-TGCTGTTGACAGTGAGCGAACAGATTAAGATTATATCTTATAGTGAAGCCACAGATGTATAAGATATAATCTTAATCTGTGTGCCTACTGCCTCGGA-3’ -
Human PSME4:
5’-TGCTGTTGACAGTGAGCGCTCAGAAGATGATACTAAGTCATAGTGAAGCCACAGATGTATGACTTAGTATCATCTTCTGAATGCCTACTGCCTCGGA-3’
Real-time quantitative polymerase chain reaction
Total mRNA was extracted with TRIzol (Invitrogen). cDNA was synthesized by use of random hexamer (M-MLV reverse transcriptase, Invitrogen). Quantitative real-time PCR was carried out by using QuantiTect SYBR Green kits (Qiagen) with a Rotor-Gene Q (Qiagen). PCR analysis was performed in triplicate and the average was regarded as a single result. The relative contents of mRNA transcripts were determined using the ddCt method, with GAPDH as the internal control. Primer sets for human YAP1 and PSME4 were purchased (Bioneer, P245469 V for YAP1 and P208885V for PSME4). Specific oligomer sets designed were as follows:
-
Human GAPDH,
sense: 5’-GTCTCCTCTGACTTCAACAGCG-3',antisense: 5’-ACCACCCTGTTGCTGTAGCCAA-3' -
Human NKX2.5
sense: 5’-AAGTGTGCGTCTGCCTTTCCCG-3',antisense: 5’-TTGTCCGCCTCTGTCTTCTCCA-3' -
Human TNNI3
sense: 5’-CGTGTGGACAAGGTGGATGAAG-3',antisense: 5’-GCCGCTTAAACTTGCCTCGAAG-3' -
Human GATA4
sense: 5’-GCGGTGCTTCCAGCAACTCCA-3',antisense: 5’-GACATCGCACTGACTGAGAACG-3' -
Mouse Gapdh,
sense: 5’-GGGTGTGAACCACGAGAAATA-3’,antisense: 5’-GTCATGAGCCCTTCCACAAT-3’ -
Mouse Nkx2.5,
sense: 5’-AAGTGCTCTCCTGCTTTCC-3’,antisense: 5’-CATCCGTCTCGGCTTTGT-3’ -
Mouse Tnni3,
sense: 5’-AGATTGCGAAGCAGGAGATG-3’,antisense: 5’-AGCCCATCCAACTCCAAAG-3’ -
Mouse Gata4,
sense: 5’-GGAAGCCCAAGAACCTGAATA-3’,antisense: 5’-CTAGTGGCATTGCTGGAGTTA-3’
Statistics
The statistical analysis was performed using PASW Statistics 27 (IBM Corp.). To compare the means of two independent groups, an independent samples t-test was conducted. For more than two groups, one-way analysis of variance (ANOVA) with post-hoc multiple comparison was used for data with one main effect, while two-way ANOVA was used for data with two or more main effects. If a significant interaction of main effects was found, further stratification was carried out to perform pairwise comparison. For post-hoc tests, Tukey’s HSD (honestly significant difference) was applied for multiple comparisons assuming equal variances. The level of statistical significance was set at p < 0.05.
RESULTS
Loss of PSME4 retards the cardiac commitment of MSCs
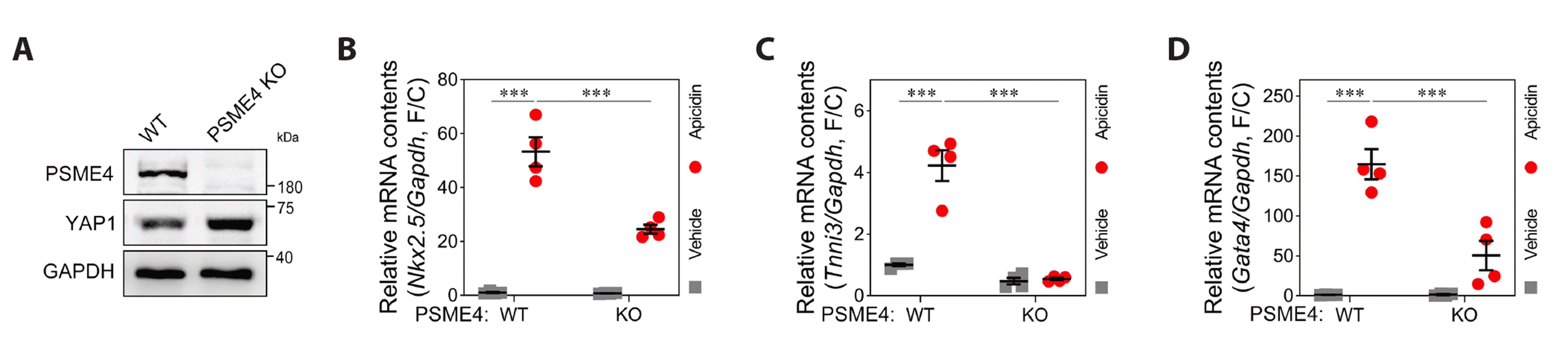
Although we demonstrated that nuclear PSME4 actively degraded acetyl-YAP1 in the nucleus in response to apicidin treatment [14], the fundamental role of PSME4 in the cardiac commitment of MSCs remains unclear. We isolated bone-marrow MSCs (BM-MSCs) from either Psme4 knock-out (KO) mice or wild-type littermates and tested the cellular characteristics of each genotypes. The Psme4 was absent in the BM-MSCs from Psme4 KO mice, and Yap1 protein was relatively abundant in those cells (Fig. 1A). After two passages for stabilization of BM-MSCs, the property of cardiac commitment was assessed in response to apicidin treatment. The measurement of the expression amount of NK2 homeobox 5 (NKX2.5), cardiac type troponin I3 (TNNI3), and GATA Binding Protein 4 (GATA4) was utilized as specific markers to represent cardiac commitment.
Overnight incubation with apicidin successfully accelerated the cardiac commitment of BM-MSCs from wild-type mice, whereas the cardiac commitment of BM-MSCs from Psme4 KO mice was significantly attenuated (Fig. 1B–D). When considering previous results [14,23], we assumed that the retardation of clearing of Yap1 in the nucleus was the responsible mechanism for delayed cardiac commitment. To answer the fundamental question, we employed the lentivirus-driven shRNA system in hTERT-MSCs.
SGEP-driven constitutive overexpression of shRNA against PSME4 reduced endogenous mRNA of PSME4 two days after infection (Fig. 2A). Like previous results (Fig. 1) obtained from BM-MSCs from Psme4 KO, targeted reduction of PSME4 decelerated cardiac commitment by apicidin treatment in hTERT-MSCs (Fig. 2B–D). We further checked the dynamics of YAP1 in response to apicidin treatment in the presence of shRNA against PSME4. YAP1 predominantly localized in the cytoplasm as reported previously, and most of the YAP1 was reduced after treatment with apicidin (Fig. 2E, 1st and 2nd panels). A notable amount of YAP1, however, was observed when PSME4 was removed (Fig. 2E, 3rd panel). Furthermore, YAP1 shuttled into the nucleus and remained for a longer duration in spite of apicidin treatment (Fig. 2E, 4th panel). Western blot analysis supported the observations acquired in Fig. 2E. After apicidin treatment, YAP1 protein expression persisted (Fig. 2F), despite a significant decrease in its mRNA levels (Fig. 2G). These findings provide further evidence for the active degradation of YAP1 protein by PSME4 in the nucleus. Taken together with previous observations, PSME4 orchestrated cardiac commitment which was induced by treatment of apicidin.
The absence of YAP1 is a mandatory step for cardiac commitment of MSCs
Next, we questioned whether the clearance of YAP1 is an essential step for cardiac commitment of MSCs. We cloned shRNA against YAP1 in a tetracycline-dependent activation system for fine regulation of shRNA and confirmed dynamic controls of shRNA expression against YAP1 (Fig. 3A). As expected, apicidin actively reduced YAP1. YAP1 shRNA itself also cleared YAP1 protein, but simultaneous treatment of both apicidin and YAP1 shRNA greatly accelerated the YAP1 clearance in the cells (Fig. 3B).
According to previous results, the cardiac commitment of MSCs was dependent on the clearance of YAP1. Hence, we postulated that simultaneous treatment of apicidin and YAP1 shRNA would potentiate cardiac commitment. To prove our hypothesis, we treated doxycycline for the induction of shRNA against YAP1 and apicidin to promote cardiac commitment. As desired, apicidin administration successfully induced cardiac commitment (1st vs. 3rd lane in Fig. 3C–E). Interestingly, cardiac commitment was greatly augmented when doxycycline was combined with apicidin treatment (3rd vs. 4th lane in Fig. 3C–E). Taken together, we could conclude that the clearance velocity of YAP1 positively correlated with cardiac commitment of MSCs.
To consolidate our tentative interpretation, we challenged the compensation of YAP1 assay after removing endogenous YAP1. Unlike the universal activity of the T7 promoter, the YAP1 promoter was greatly suppressed by treatment of apicidin through transactivation of p21 [24,25]. This discrepancy in response to apicidin allowed for the differential regulation of exogenous YAP1 protein. We transfected a plasmid carrying the wild type of YAP1 or an acetylation-dead mutant of YAP1 that was tagged with Flag. The total protein amount of the mixture of both endogenous and exogenous YAP1 was almost similar (Fig. 4A), but apicidin preferentially degraded both the endogenous YAP1 and the wild type flag-tagged YAP1, whereas the acetylation-resistant mutant of YAP1 allowed for a prolonged half-life (Fig. 4B).
As shown in Fig. 3, the simultaneous incubation of doxycycline and apicidin greatly increased the cardiac commitment of MSCs when compared to the apicidin-treatment alone condition (1st vs. 2nd lanes in Fig. 4C–E). Overexpression of the wild type of YAP1, however, still potentiated the cardiac commitment (3rd and 4th lanes in Fig. 4C–E), but the overall activity was significantly retarded in the presence of YAP1 protein (2nd vs. 4th lanes in Fig. 4C–E). Most strikingly, the abundance of acetylation-dead YAP1 failed to accelerate the cardiac commitment of MSCs (5th vs. 6th or 2nd vs. 6th lanes in Fig. 4C–E). When combined with previous results, we could conclude that the fine regulation of YAP1, the transient existence of nuclear YAP1, and the subsequent clearance by PSME4 were mandatory for the cardiac commitment of MSCs.
HDAC inhibition commonly prompts cardiac commitment
We have delineated that apicidin successfully provoked the cardiac commitment of MSCs. However, we asked whether the cardiac commitment of MSCs by apicidin was due to the common HDAC inhibition effect or the unique features of apicidin. We selected tubastatin A, a cytosolic HDAC inhibitor, and assessed the cardiac commitment in the presence of tubastatin A. A relatively lower dose of tubastatin A induced cardiac commitment in a mild manner, and a higher dose of tubastatin A promoted the cardiac commitment of MSCs as strongly as the treatment of apicidin (Fig. 5A–C). A considerable amount of YAP1 protein was reduced by tubastatin A treatment, which was comparable to that of apicidin treatment (Fig. 5D).
Tubastatin A is a well-known cytosolic HDAC inhibitor that targets HDAC6 and HDAC10, while apicidin is a potent class I HDAC inhibitor and weakly acetylated cytosolic tubulin [14]. Hence, we further tested the effect of HDAC6 inhibition on cardiac commitment. We transfected siRNA against either HDAC6 or non-targeting siRNA in hTERT-MSCs. Two days of siRNA treatment successfully reduced both endogenous HDAC6 mRNA (Fig. 5E) and HDAC6 protein (Fig. 5F). Interestingly, down-regulation of HDAC6 significantly promoted cardiac commitment, which was comparable to the effect of treatment with apicidin or tubastatin A. Overall, we concluded that inhibition of cytosolic HDAC6 initiates cardiac commitment of MSCs.
DISCUSSION
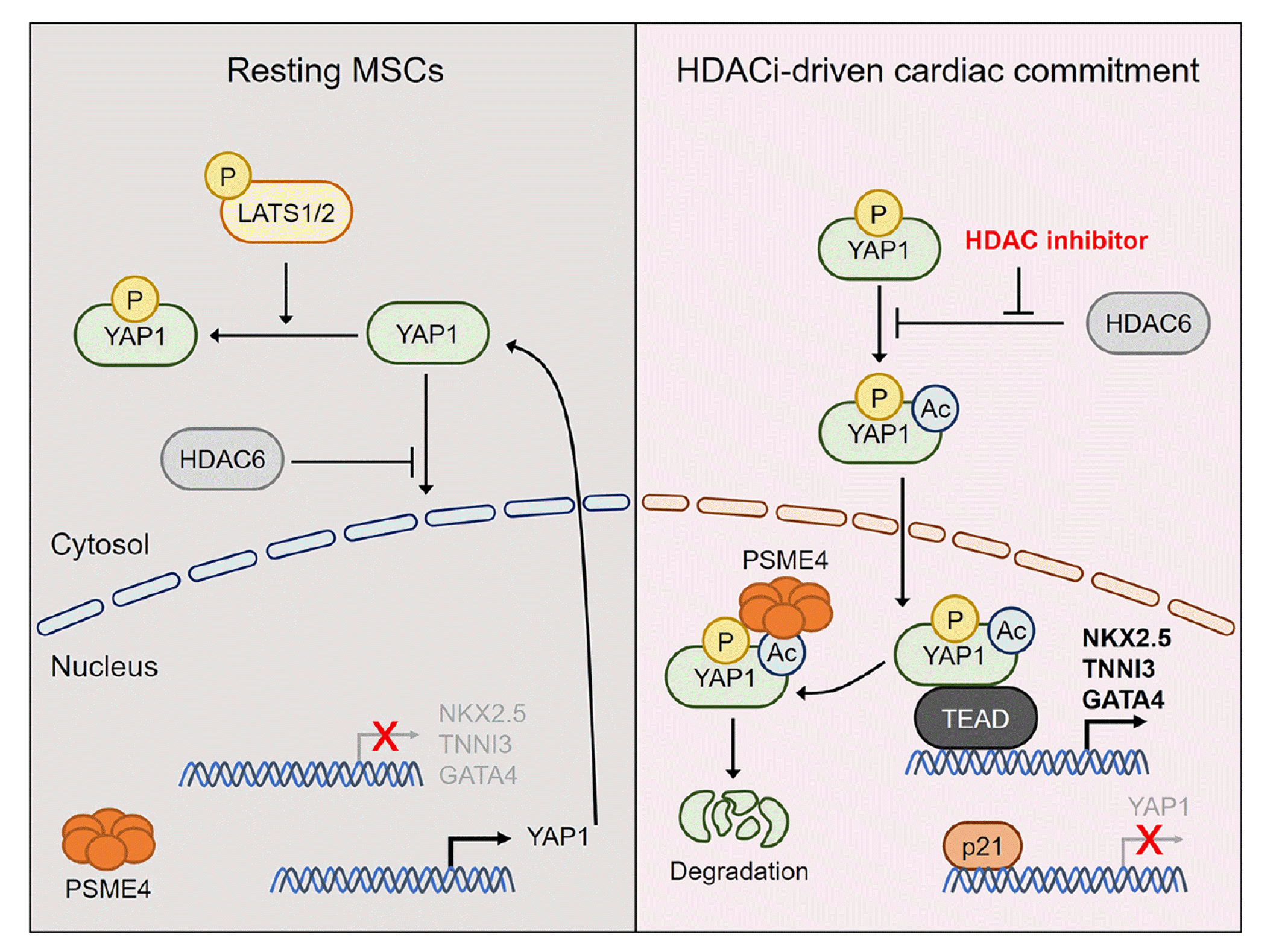
In this study, we investigated the role of PSME4 and YAP1 in cardiac commitment of MSCs. We demonstrated that the absence of PSME4 and the presence of YAP1 are both associated with the retardation of cardiac commitment. We also showed that the clearance of YAP1 is an essential step, and that inhibition of cytosolic HDAC6 initiates cardiac commitment (Fig. 6).
The role of YAP1 in stem cell differentiation and organ size regulation has been a topic of debate in recent years [26-28]. While some reports have concluded that YAP1 inhibits stem cell differentiation [28], others have presented evidence that YAP1 accelerates stem cell differentiation [27]. Additionally, YAP1 has been shown to regulate organ size, which further complicates our understanding of its role in stem cell differentiation. One possible explanation for the discrepancy in the literature could be due to the context-dependent nature of YAP1 signaling [29]. YAP1 interacts with a variety of different transcription factors and signaling pathways, and its activity is tightly regulated by post-translational modifications, such as acetylation and phosphorylation [14,16,30-32]. Therefore, the specific downstream targets of YAP1 may vary depending on the cell type and signaling context. Another possibility is that the role of YAP1 in stem cell differentiation is stage-specific. For example, YAP1 may be necessary to maintain the stemness of undifferentiated stem cells, but once differentiation has been initiated, YAP1 may act as a negative regulator to prevent the cells from reverting back to a stem cell-like state. The conflicting results regarding the role of YAP1 in stem cell differentiation also highlight the need for more comprehensive studies to fully understand its function in this context. Future studies should aim to investigate the specific downstream targets of YAP1 and the signaling pathways involved in its regulation of stem cell differentiation. Additionally, it will be important to examine the role of YAP1 at different stages of stem cell differentiation and in different cell types to gain a more complete understanding of its function in this process.
Our results demonstrate that PSME4 is an important regulator of cardiac commitment in MSCs. The absence of PSME4 significantly attenuated cardiac commitment in response to apicidin treatment in both BM-MSCs from Psme4 KO mice (Fig. 1) and hTERT-MSCs expressing shRNA against PSME4 (Fig. 2). We previously showed that nuclear PSME4 actively degrades acetyl-YAP1 in response to apicidin treatment [14]. Therefore, we assume that the retardation of clearing of Yap1 in the nucleus was the responsible mechanism for delayed cardiac commitment. Our results suggest that PSME4 orchestrates cardiac commitment, which is induced by treatment with apicidin. Our study also highlights the crucial role of YAP1 in cardiac commitment of MSCs. The clearance of YAP1 is a mandatory step for cardiac commitment, and simultaneous treatment of apicidin and YAP1 shRNA greatly accelerated YAP1 clearance and potentiated cardiac commitment (Fig. 3). We showed that overexpression of the wild-type YAP1 protein retarded cardiac commitment, and the overall activity was blocked in the prolonged presence of YAP1 protein. This suggests that the fine regulation of YAP1, the transient existence of nuclear YAP1, and the subsequent clearance by PSME4 are all mandatory for the cardiac commitment of MSCs (Fig. 6). Our findings are consistent with previous studies that have demonstrated the importance of YAP1 in cardiac development and differentiation [22,33,34].
In addition, we investigated the role of apicidin, a HDAC inhibitor, in promoting cardiac commitment of MSCs. We examined whether the cardiac commitment induced by apicidin was due to the common HDAC inhibition effect or the unique features of apicidin (Fig. 5). Our findings showed that tubastatin A, a cytosolic HDAC inhibitor, also promotes cardiac commitment of MSCs, indicating that the inhibition of HDACs is a critical step in promoting cardiac commitment. Furthermore, we demonstrated that down-regulation of HDAC6, which is predominantly localized in the cytosol, significantly promoted cardiac commitment of MSCs, which was comparable to the effect of treatment with apicidin or tubastatin A. These results suggest that inhibition of cytosolic HDAC6 is an important mechanism for initiating cardiac commitment of MSCs. Our study sheds light on the mechanism underlying the promotion of cardiac commitment of MSCs by HDAC inhibitors and provides valuable insights for developing effective strategies for cardiac regeneration.
Overall, our study provides new insights into the molecular mechanisms underlying cardiac commitment of MSCs. The results of this study provide important insights into the role of PSME4 and YAP1 in the cardiac commitment of MSCs. The findings suggest that regulating the activity of PSME4 or YAP1 could be a promising therapeutic strategy for promoting the differentiation of MSCs into cardiomyocytes for cardiac regeneration. One possible approach could be to develop small molecules that selectively target PSME4 or YAP1, and promote their activity in promoting cardiac commitment. Another approach could involve the use of gene therapy to overexpress or knockdown PSME4 or YAP1, depending on the specific therapeutic goal. Further research is needed to fully understand the mechanisms underlying the PSME4-YAP1 axis and to identify additional molecules that can modulate this pathway. Ultimately, the development of drugs targeting PSME4 and YAP1 could offer a new avenue for treating cardiac diseases and promoting cardiac regeneration.
 XML Download
XML Download