

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Approximately 33.5 million patients worldwide had cardiac arrhythmia almost 10 years ago; the most common arrhythmia is atrial fibrillation [1,2]. Arrhythmia is produced by several mechanisms; most arrhythmias are related to ion channels and most anti-arrhythmic agents are ion channel modulators [3-5]. Anti-arrhythmic agents are classified into four types based on their ability to block ion channels or cell receptors [6]. Class I anti-arrhythmic drugs target Na+ channels and are divided into classes Ia–c according to their degree of inhibition on Na+ channels [4]. Encainide is a class Ic anti-arrhythmic drug used since the 1980s [7]. It is a safe and well-tolerated anti-arrhythmic agent, with some minor side effects such as dizziness, headache, and nausea [8,9]. However, the side effects of encainide on other ion channels, particularly the voltage-dependent K+ (Kv) channels expressed in vascular smooth muscle cells, have not been elucidated.
Smooth muscle cells of the vasculature regulate vascular tone and function. In turn, altered vascular smooth muscle cells are strongly associated with cardiovascular diseases, such as atherosclerosis, diabetes, hypertension, and hypertrophy [10-12]. Vascular contractility is regulated by ion channels expressed in smooth muscles [13]. Several ion channels have been identified on vascular smooth muscle cells. Of these, Kv channels are the most abundant in the membrane and control the vascular tone by changing the resting membrane potential [14,15]. Kv channels comprise diverse subtypes that regulate smooth muscle cell functions. Therefore, modulation of Kv subtypes is a potential therapeutic target for cardiovascular diseases. For instance, blockade of Kv1.3 affects the proliferation of smooth muscle cells and is closely related to the pathogenesis of atherosclerosis [16,17].
Considering the therapeutic benefit of encainide for cardiac arrhythmia and the pathophysiological importance of vascular Kv channels, it is essential to explore the inhibitory effects of encainide on vascular Kv channels to avoid miscomprehension of vascular function data and to prevent side effects on vasculature. In the present study, we demonstrated the inhibitory effect of encainide on vascular Kv channels using freshly segregated rabbit coronary arterial smooth muscle cells. We found that encainide inhibited vascular Kv channels in a concentration-dependent and use-independent manner. This inhibitory reaction occurs by changing the gating property of the channel.
METHODS
Single smooth muscle cell isolation
Male New Zealand White rabbits (2.2–2.4 kg) were sacrificed by instantaneous injection of heparin (120 U/kg) and sodium pentobarbital (35 mg/kg) into the ear vein. All animal experiments followed the National Institutes of Health guidelines and were approved by the Animal Ethics Committee of the Yangzhou University (approval no.: YZU220180489). The heart was immediately separated and the left descending coronary artery in the heart was segregated for single cell acquisition. Adipose tissues were removed using normal Tyrode’s solution under a stereomicroscope. The endothelial cells were eliminated by injecting air bubbles into the arterial lumen. For single cell acquisition, the arteries were treated using two-step enzymatic procedures. First, the arteries were rinsed in 1 ml of Ca2+-removed normal Tyrode’s solution containing papain (0.8 mg/ml), dithiothreitol (DTT, 1.2 mg/ml), and bovine serum albumin (BSA, 1.1 mg/ml), and incubated for 22 min at 37°C–38°C. Second, the arteries were rinsed into 1 ml of Ca2+-removed normal Tyrode’s solution containing collagenase (2.4 mg/ml), DTT (1.2 mg/ml), and BSA (1.1 mg/ml) for 20–23 min at 37°C–38°C. Fresh cells were separated by mechanical agitation in Kraft–Brühe solution using a heat-polished Pasteur pipette. The freshly separated single cells were kept at 4°C–6°C and used within a day.
Solutions and chemicals
The composition (mM) of normal Tyrode’s solution was KCl, 5.4; NaCl, 135; CaCl2, 1.7; MgCl2, 1.2; NaH2PO4, 0.37; glucose, 15; and HEPES, 5. The pH of the solution was adjusted to 7.4 using NaOH. The composition (mM) of the Kraft–Brühe solution was KOH, 71; KCl, 56; L-glutamate, 53; KH2PO4, 21; taurine, 18; MgCl2, 1.9; glucose, 15; EGTA, 0.5; and HEPES, 10. The pH of the solution was adjusted to 7.3 using KOH. The composition (mM) of the pipette solution for recording of Kv channels was NaCl, 5.5; K-aspartate, 113; KCl, 28; MgCl2, 1.6; Mg-ATP, 4; EGTA, 10; and HEPES, 8. The pH of the solution was adjusted to 7.25 using KOH. Encainide (Sigma-Aldrich) was dissolved in ethanol. Guangxitoxin and DPO-1 (Tocris Cookson) were dissolved in dimethyl sulfoxide. The final solvent contents were below 0.1%, which did not alter the Kv currents.
Electrophysiology and data analysis
Kv currents from single vascular smooth muscle cells were measured using an EPC-10 amplifier (HEKA Elektronik GmbH). Patchpro software was applied for protocol generation and data acquisition. The patch electrodes were pulled using a P-97 microelectrode puller (Sutter Instrument) using a thin glass electrode. The patch electrode had a resistance of 2.5–3.5 MΩ when filled with the pipette solution. The average cell capacitance was 16.22 ± 1.33 pF (n = 15). The perforated-patch clamp technique was applied to record the resting membrane potential. Nystatin (100 µg/ml) was included in the internal solution. The success of the perforated-patch configuration was confirmed by the observation of a slow capacitive current. All experiments were performed at room temperature.
Origin 8.0 software (Microcal Software, Inc.) was used for data analysis. The kinetics of drug–ion channel interaction was described using a first-order blocking scheme. The half (mid)-maximal inhibitory (IC50) value and the Hill coefficient (n) were determined by analysis of concentration-dependent data and fitted to the following Hill equation:
where ƒ is the relative current inhibition (ƒ = 1 – Idrug / Icontrol) at the given potential and [D] is the encainide concentration.
Steady state activation curves were constructed using tail currents, which were produced by returning the voltage to –40 mV after application of short depolarizing voltages (20–50 ms) from –80 to +60 mV in 10-mV increments. The elicited tail currents at each depolarization voltage were normalized to the maximal tail current. The activation curves were fitted using the Boltzmann equation given below:
where V1/2 is the half (mid)-maximal activation voltage, V is the test voltage, and k is the slope value.
The steady-state inactivation curves were constructed by applying the given voltage to +40 mV at a duration of 600 ms after 7-s preconditioning voltages from −80 to +30 mV in 10-mV increments. Steady state inactivation curves were fitted with below Boltzmann equation:
where V1/2 is the half (mid)-maximal inactivation voltage, V is the preconditioning voltage, and k is the slope value.
RESULTS
Encainide-induced inhibition of Kv currents in rabbit coronary arterial smooth muscle cells
Encainide-induced inhibition of Kv current was recorded in freshly isolated rabbit coronary arterial smooth muscle cells using the whole-cell patch clamp technique. To exclude interference by other K+ currents, EGTA (10 mM) and ATP (4 mM) were added to the internal solution to inhibit large-conductance Ca2+-activated K+ (BKCa) and ATP-dependent K+ (KATP) channels, respectively. The use of large coronary arteries prevents interference from the inward rectifier K+ (Kir) channel because this channel is only expressed in small vessels (microvessels). Kv current was recorded using 600-ms depolarizing steps from –80 to +60 mV with a holding potential of –80 mV in steps of 10 mV. Control Kv current was evoked rapidly after voltage stimulation and decreased partially due to intrinsic inactivation (Fig. 1A). Application of 10 μM encainide immediately (within 1 min) reduced Kv currents (Fig. 1B); this effect was partially washed out (by almost 50%). Current-voltage (I-V) relationships of steady-state current are shown in Fig. 1C. At +60 mV, application of 10 μM encainide blocked the Kv current by 46.85%. The degree of encainide-induced inhibition was also similarly observed in femoral and mesenteric arterial smooth muscle cells (Supplementary Fig. 1). In addition, application of 10 μM encainide had no effect on the BKCa and KATP currents (Supplementary Fig. 2).
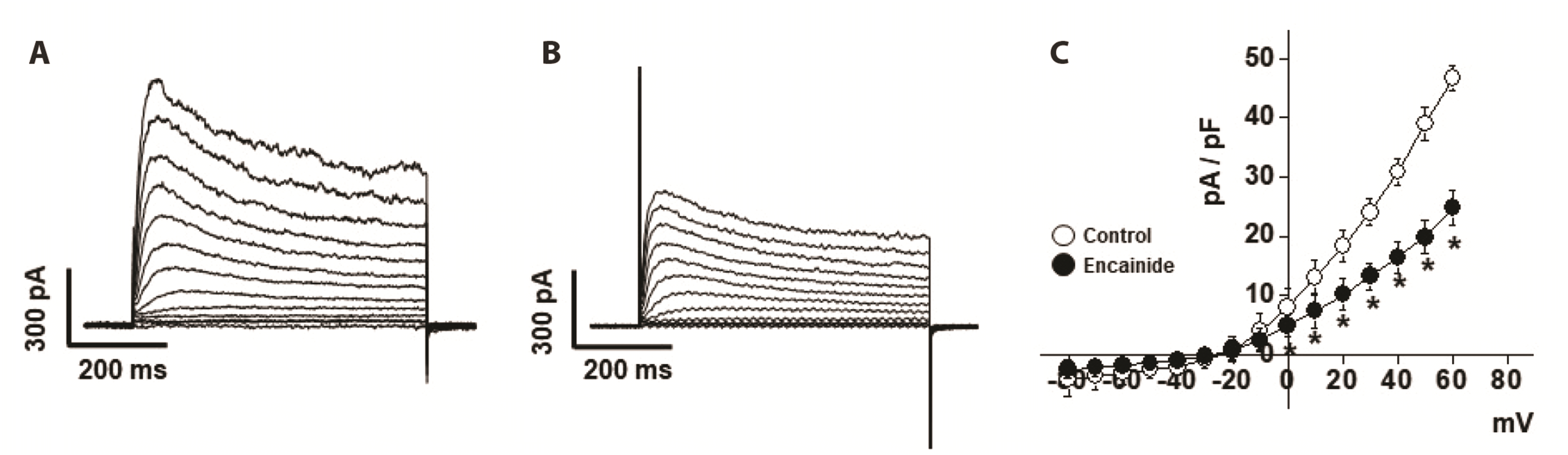
Fig. 1
Effects of encainide on voltage-dependent K+ (Kv) currents in rabbit coronary arterial smooth muscle cells.
The Kv currents were recorded using a 600-ms depolarizing pulse from −80 to +60 mV in steps of 10 mV at a holding potential of −80 mV under the control condition (A) and in the presence of 10 μM encainide (B). (C) Summary of the current voltage (I-V) relationship at steady-state Kv currents in the control condition (○) and in the presence of 10 μM encainide (●). n = 6. All n means the number of cells. Only one cell was examined from a rabbit to minimize individual differences. *p < 0.05 (control vs. encainide, at each voltage).
![]()
Encainide inhibits Kv currents in a concentration-dependent manner
To determine whether the inhibitory effect of encainide on Kv currents increases in a concentration-dependent manner, several concentrations of encainide (0.1, 0.3, 1, 3, 10, 30, and 100 μM) were sequentially administered to the Kv current. Kv current was recorded using single-step depolarizing voltages to +60 mV from a holding potential of −80 mV. Fig. 2A shows that inhibition of the Kv currents were gradually increased as the encainide concentration increased. Using the Hill equation, analysis of the concentration-dependent inhibition of the Kv channels by encainide provided an IC50 value of 8.9 ± 1.75 μM and a slope value of 0.7 ± 0.06 (Fig. 2B). These results indicate that encainide inhibited the Kv current in a concentration-dependent manner.
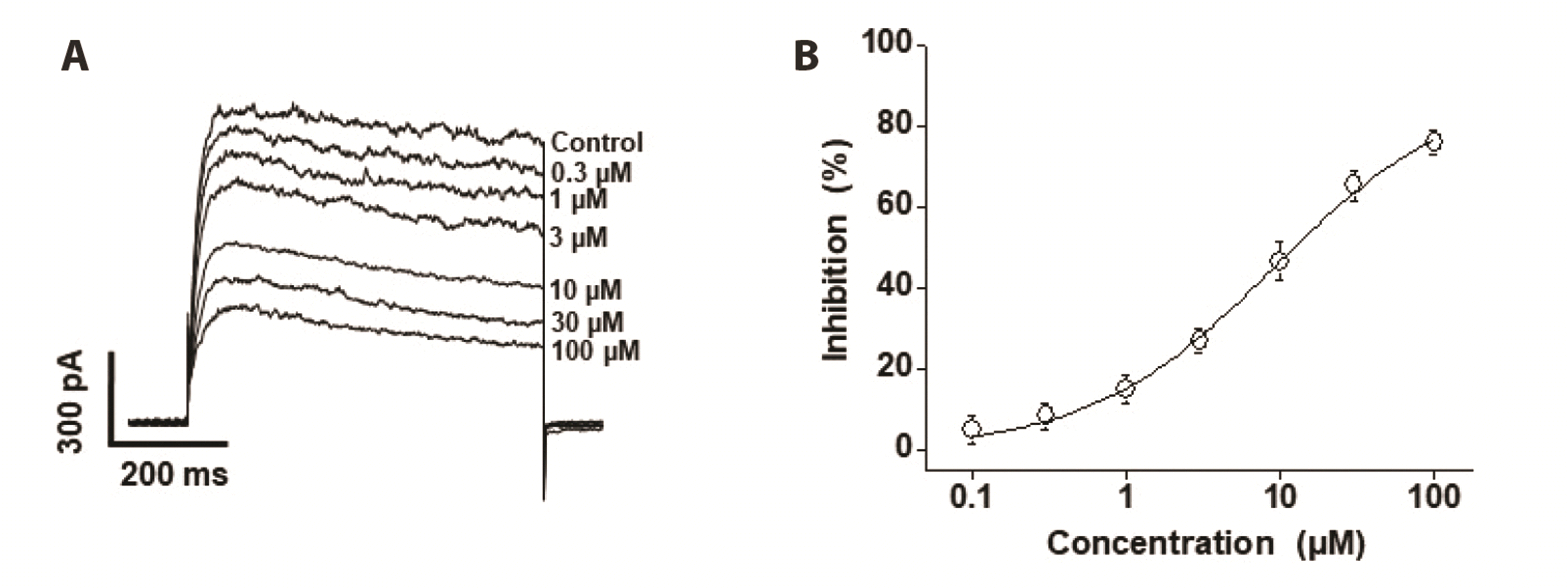
Fig. 2
Concentration-dependent inhibition of the voltage-dependent K+ (Kv) channel by encainide.
(A) Representative current traces were elicited by 600-ms depolarizing pulses from a holding potential of −80 mV in the presence of 0, 0.1, 0.3, 1, 3, 10, 30, and 100 μM encainide. (B) Concentration-dependent curve for the inhibitory effect of encainide on the Kv current was measured at steady-state (○) and normalized to the current amplitude observed in the absence of encainide (control). Normalized currents were fitted using the Hill equation. n = 6 for all.
![]()
Effects of encainide on steady state activation and inactivation of Kv channels
To determine whether encainide-induced inhibition of Kv channels was induced by alteration of channel gating properties, we tested the effects of encainide on steady-state activation and inactivation curves. The steady-state activation curve was drawn based on analysis of tail currents and the results were fitted using the Boltzmann function, as described in the Methods section. Fig. 3A shows that the application of encainide shifted the steady-state activation curves to the right. The mid-point of the activation kinetics (V1/2) and the slope factor (k) were −10.96 ± 0.82 mV and 14.42 ± 0.76 under the control condition, and 2.34 ± 1.02 mV and 16.91 ± 1.05 in the presence of encainide, respectively.
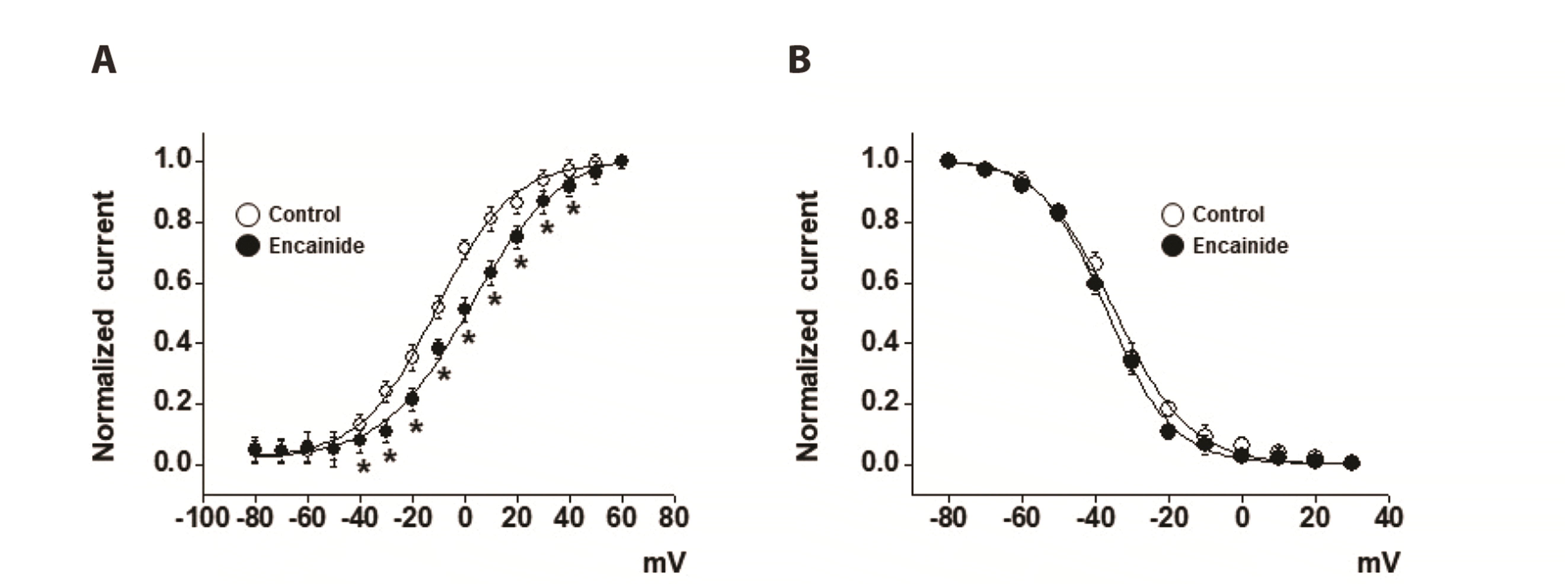
Fig. 3
Influence of encainide on steady-state activation and inactivation curves.
(A) Activation curves under the control conditions (○) and in the presence of 10 μM encainide (●). Activation curves were drawn based on analysis of tail currents, which were induced by applying a short depolarizing step from −80 to +60 mV in 10-mV increments at an −80 mV holding potential, followed by a return potential of −40 mV. The acquired tail currents were normalized to the maximum value of the tail current. n = 6. *p < 0.05 (control vs. encainide, at each voltage). (B) Inactivation curves under the control conditions (○) and in the presence of 10 μM encainide (●). Inactivation curves were obtained by applying a test step to +40 mV after 7-s pre-conditioning at different voltages. The currents induced by the test step were normalized to the peak amplitude of the pre-conditioning step-induced currents. n = 5.
![]()
The steady-state inactivation curve was drawn by analyzing the peak amplitudes of a typical pulse protocol; the results were fitted using another Boltzmann function, as described in the Methods section. As shown in Fig. 3B, application of encainide did not shift the steady-state inactivation curves. The half-point of the inactivation kinetics (V1/2) and the slope factor (k) were −34.74 ± 0.77 mV and 10.22 ± 0.60 under control condition, and −36.28 ± 0.51 mV and 9.20 ± 0.59 in the presence of encainide, respectively.
Use dependence of Kv channel inhibition by encainide
To examine whether the encainide-induced decrease of Kv channels is use (state)-dependent, we recorded Kv currents by administering 20 repeated 150-ms depolarizing pulses to +60 mV from a holding potential of −80 mV at 1 and 2 Hz frequencies. Under control conditions, the administration of 20 repetitive pulses decreased the amplitudes of the Kv channel currents by 12% and 23% at 1 and 2 Hz, respectively. However, application of 10 μM encainide did not alter the degree of Kv current inhibition at 1 and 2 Hz (Fig. 4A and 4B), suggesting that encainide inhibits the Kv current in a use (state)-independent manner.
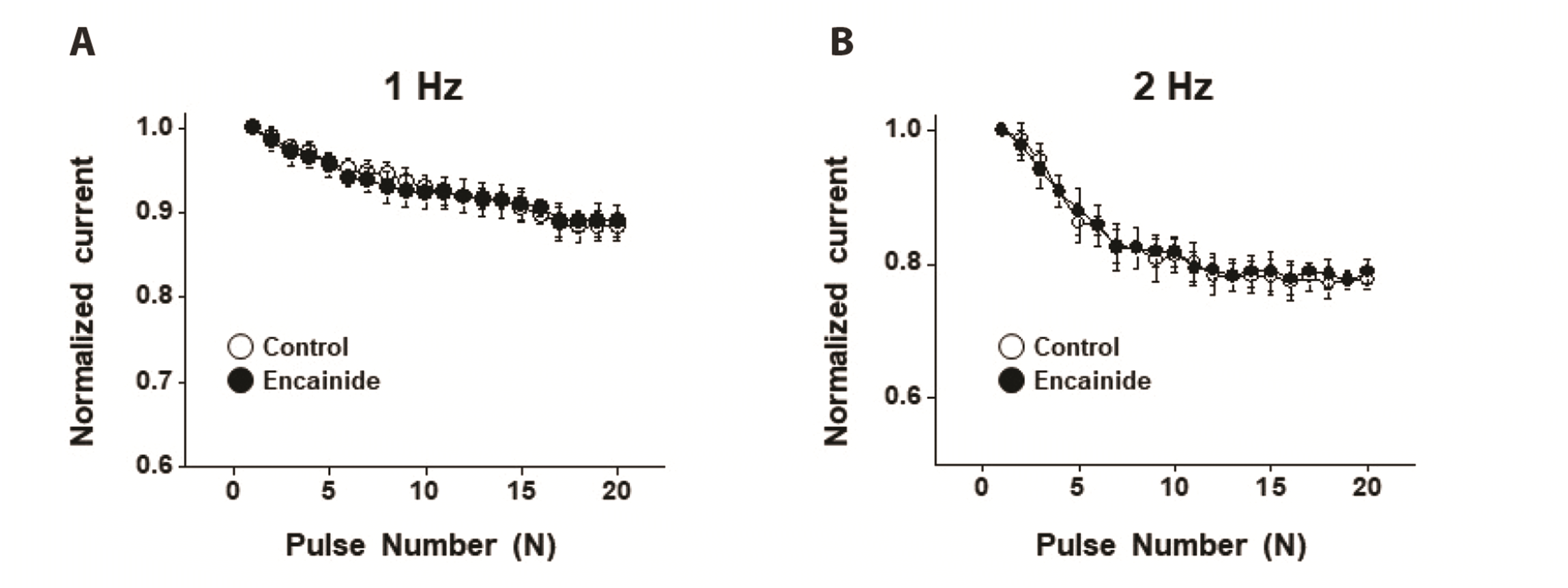
Fig. 4
Use (state)-independent effect of encainide on voltage-dependent K+ currents.
Twenty repeated depolarizing pulses were applied from a holding potential of −80 mV to +60 mV at frequencies of 1 (A) and 2 (B) Hz in the absence (○) and presence (●) of 10 μM encainide The peak currents were normalized using the peak current recorded after the first pulse and plotted against the pulse number. n = 6.
![]()
Effect of encainide on Kv currents in the presence of Kv1.5 and Kv2.1 subtype inhibitors
To evaluate the role of the Kv subtype in encainide-induced decrease of Kv channels, we pre-treated the channels with Kv1.5 or Kv2.1 subtype inhibitor prior to the application of encainide. As shown in Fig. 5A, the Kv1.5 subtype inhibitor DPO-1 (1 μM) rapidly inhibited the Kv current. Additional treatment of encainide slightly inhibited the Kv current, albeit without statistical significance (Fig. 5B). Pretreatment with the Kv2.1 subtype inhibitor guangxitoxin (100 nM) also effectively decreased the Kv current (Fig. 5C). However, additional treatment with encainide blocked the Kv current to a level similar to that observed with encainide alone (Fig. 5D), suggesting that the inhibitory effect of encainide on Kv channels involved the Kv1.5 subtype, but not the Kv2.1 subtype.
Fig. 5
Roles of Kv1.5 and Kv2.1 subtypes in encainide-induced inhibition of Kv channels.
Representative Kv currents were recorded using 600-ms depolarizing steps of +60 mV from a holding potential of −80 mV. (A) Superimposed currents under control conditions and in the presence of 1 μM DPO-1 or 1 μM DPO-1 + 10 μM encainide. (B) Summary of the results shown in panel (A). n = 5. NS, not significant (DPO-1 vs. DPO-1 + encainide). (C) Superimposed currents under control conditions and in the presence of 100 nM guangxitoxin or 100 nM guangxitoxin + 10 μM encainide. (D) Summary of the results shown in panel (C). n = 5. *p < 0.05 (guangxitoxin vs. guangxitoxin + encainide). Kv, voltage-dependent K+.
![]()
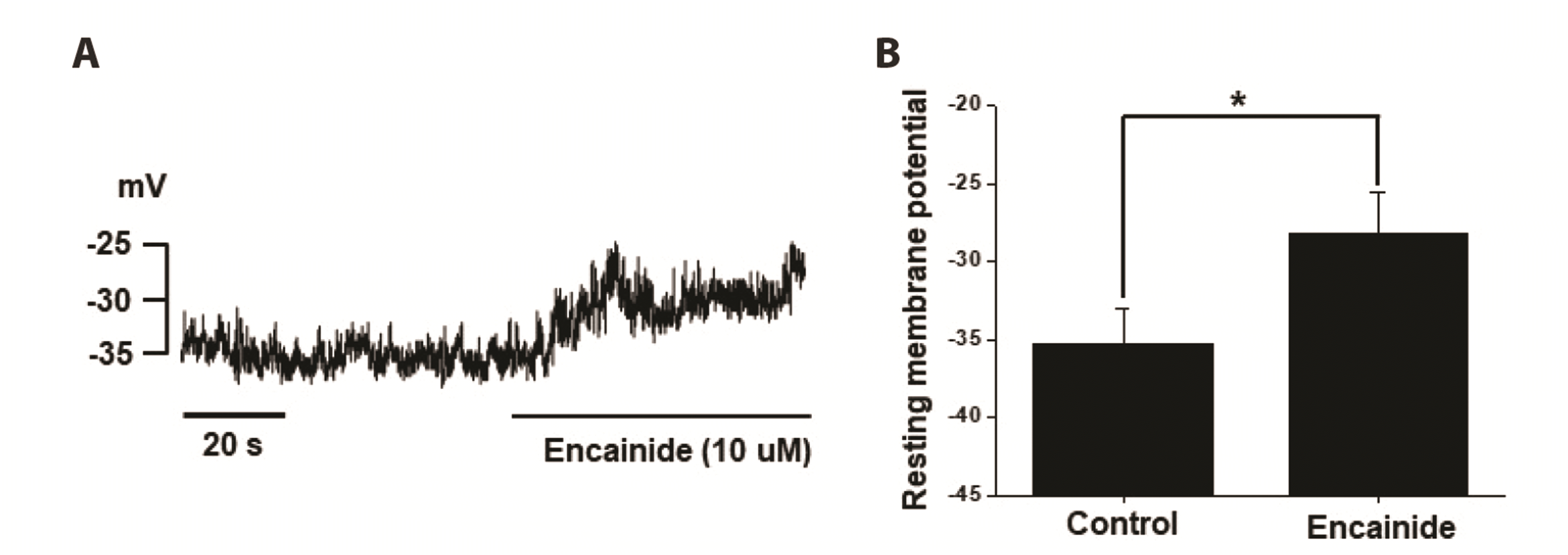
Effect of encainide on resting membrane potential
DISCUSSION
We investigated the inhibitory effect of encainide on Kv channels in smooth muscle cells from rabbit coronary arteries. Our results suggest that encainide inhibits Kv channels by shifting the steady-state activation curve in a dose-dependent and use (state)-independent manner. We found that encainide inhibits Kv channels on vascular smooth muscle regardless of its anti-arrhythmic effect. First, the response time of encainide on inhibition of Kv current is very short (within 1 min), suggesting direct inhibition of Kv channels by encainide. Second, encainide effectively shifted the steady-state activation curve toward a more positive potential (Fig. 3A), suggesting that encainide inhibited Kv channels by direct interaction with voltage sensors, inducing changes in channel structure. Third, vascular smooth muscle cells, which are non-excitable cells, do not express Na+ channels. Although the main target of the anti-arrhythmic effect of encainide is the Na+ channel, the inhibitory effect of encainide on Kv channels is independent of Na+ channels. Therefore, the inhibitory effect of encainide on Kv currents was not related to Na+ channel inhibition and was attributed to direct interaction with Kv channels.
Arrhythmia, a common heart disease, is a crucial cause of sudden cardiac death [1,2]. In the modern era, insufficient sleep, smoking, and excessive alcohol consumption are closely related to arrhythmias [1]. Surgical and pharmacological treatments are widely used for arrhythmia treatment. In the 1970s, anti-arrhythmic drugs were categorized into classes I–IV by Vaughan Williams [6]. The main target of class I antiarrhythmic drugs is the Na+ channel. These drugs were further classified into classes Ia–c according to their effect on action potential duration. Class Ic drugs do not alter action potential duration and have the strongest effect on the initiation phase 0 of depolarization [4]. To date, several class Ic anti-arrhythmic drugs, such as flecainide, propafenone, moricizine, lorcainide, and encainide, have been approved for use. These drugs have common noncardiac side effects. Similar to other class Ic antiarrhythmic drugs, encainide has side effects such as nausea, dizziness, headache, and taste disorders [8,9]. However, the side effects of encainide due to its effects on other ion channels are unclear. In the present study, we investigated the effects of encainide on Kv channels in coronary arterial smooth muscle cells. We found that encainide inhibits coronary Kv channels and cardiac Na+ channels. Therefore, the effects of encainide on cardiac and vascular smooth muscle cells should be considered when prescribing encainide as an anti-arrhythmic agent, particularly in patients with hypertension.
Vascular smooth muscle cells have abundant potassium channels, such as BKCa, KATP, Kir, and Kv channels. Of these, Kv channels, which are the most abundantly expressed K+ channels in vascular smooth muscle, regulate proliferation, migration, and vascular tone by changing the membrane potential of vascular smooth muscle cells [14,15]. Additionally, changes in vascular Kv channels are closely associated with various cardiovascular diseases, such as diabetes, atherosclerosis, hypertension, and abnormal blood pressure [10-12]. Therefore, functional recovery of Kv channels is a crucial therapeutic target for cardiovascular diseases. The unexpected effects of certain drugs such as class Ic anti-arrhythmic drugs on vascular Kv channels should be comprehensively explored because of the pathophysiological importance of Kv channels. The class Ic anti-arrhythmic drug propafenone inhibits vascular Kv channels in a dose-dependent manner [18]. However, unlike encainide, propafenone has no significant effect on the steady-state activation and inactivation curves. Furthermore, propafenone-induced inhibition of Kv currents occurs in a use-dependent manner. Another class Ic anti-arrhythmic drug, flecainide, inhibits vascular Kv channels. Similar to encainide, flecainide inhibits Kv channels in a dose-dependent but not use-dependent manner. Additionally, flecainide shifted the steady-state inactivation curve, but not the activation curve [19]. Recently, the class Ic anti-arrhythmic drug lorcainide has been shown to have an inhibitory effect. Lorcainide inhibits arterial Kv channels in a concentration- and use-dependent manner by altering their voltage sensor for inactivation gating [20]. The class Ic anti-arrhythmic drugs have different inhibitory effects on vascular smooth muscle cells. Although the exact reason is unknown, it may be due to structural differences among class Ic anti-arrhythmic drugs. Further studies are needed to confirm this.
To date, various subtypes of Kv channels have been identified in vascular smooth muscle cells, including Kv1.1, Kv1.2, Kv1.4, Kv1.5, Kv2.1, Kv7, and Kv9.3 subtypes [14,15]. However, the expression of vascular Kv subtypes differs among species. Most studies of Kv subtype expression have been conducted in rat, mouse, and human arteries [21]. Therefore, it is unclear which Kv subtypes are expressed in rabbit arteries. However, the Kv subtypes commonly expressed in most species are the vascular Kv1.5 and Kv2.1 subtypes [22], and specific inhibitors of these subtypes have been developed. Therefore, the roles of Kv1.5 and Kv2.1 subtypes in encainide-induced inhibition of Kv channels were investigated. Our results showed that the application of a Kv1.5 subtype or Kv2.1 subtype inhibitor effectively inhibited the Kv currents, suggesting that these subtypes are expressed in rabbit coronary arterials smooth muscle cells (Fig. 5). Additionally, we identified a role of the Kv1.5 subtype in encainide-induced inhibition of Kv channels. Due to limitations of native cell experiments, involvement of other Kv subtypes could not be evaluated in the present study. Further studies are needed to determine the roles of specific subtypes in the effects of encainide using an expression system for specific Kv subtypes.
Encainide is administered at a dose of 50 mg/day. After oral administration, encainide is rapidly absorbed and reaches a peak plasma concentration of 511 nM [23]. Our results show that encainide inhibited Kv channels with an IC50 value of 8.9 ± 1.75 μM, which was higher than the peak plasma concentration. However, application of 300 nM encainide effectively inhibited the Kv current (Fig. 2). Considering that arterial smooth muscle cells have high input resistance, small alterations in ionic conductance alter the vascular tone. Furthermore, inadvertent drug use may increase the blood concentration of encainide. Therefore, the dose of encainide should be carefully selected, particularly for patients with multiple cardiovascular diseases.
In summary, our study demonstrated the inhibitory effects of encainide on Kv channels using native rabbit coronary arterial smooth muscle cells. Encainide inhibited vascular Kv channels in a dose-dependent and use (state)-independent manner by altering the steady-state activation curve.
SUPPLEMENTARY MATERIALS
Supplementary data including two figures can be found with this article online at https://doi.org/10.4196/kjpp.2023.27.4.399.
 XML Download
XML Download