

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is a metabolic syndrome in chronic liver disease, affecting > 25% of the adult population worldwide [1]. Obesity is a major cause of NAFLD and other metabolic diseases [2]. Various factors contribute to NAFLD development, with obesity and type 2 diabetes mellitus (T2DM) being the most potent factors leading to NAFLD via dysregulation of glucose metabolism and insulin resistance (IR) [3]. Dysfunction in glucose uptake, IR, and hyperlipidemia can induce hepatocellular apoptosis by triggering inflammation or disrupting cellular homeostasis [4]. Although various drugs have been developed to target inflammation, oxidative stress, lipid accumulation, and IR, they exert adverse side effects in patients with NAFLD [5].
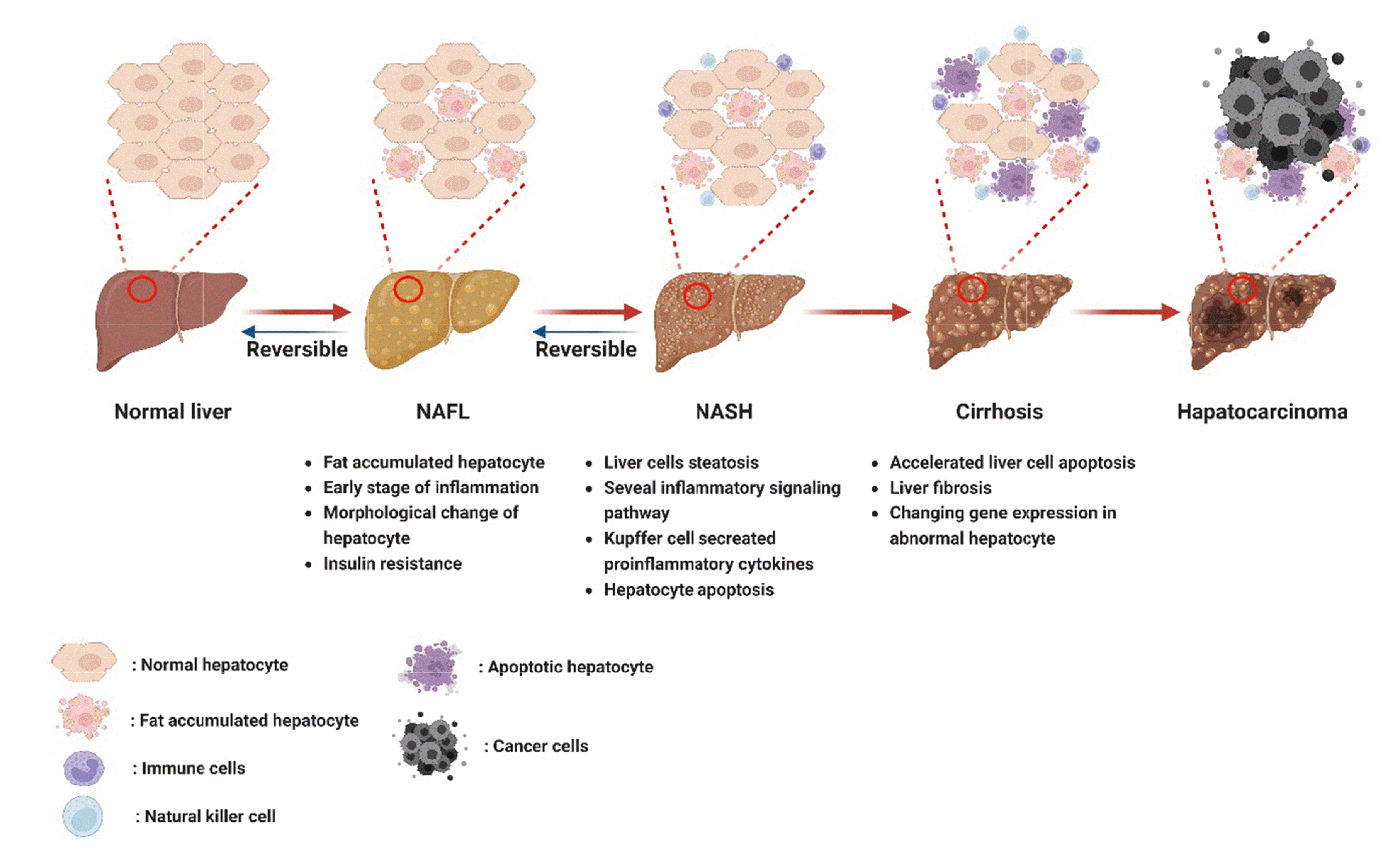
Liver disease progression involves hepatic steatosis, non-alcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma (HCC) [4]. NAFLD pathogenesis can be explained by the “multiple-hit theory,” which elucidates the progression from normal liver phenotype to HCC based on each stage [6]. Multiple-hit theory suggests that fat accumulation, IR, neoadipogenesis, and hyperglycemia due to excess weight, obesity, or excess calories leads to a change from a normal liver phenotype to NAFLD [6]. Oxidative stress, chronic inflammation, mitochondrial dysfunction, and fibrosis induce the development of NASH [4]. In addition, disruption of the gut microbiota, mitochondrial dysfunction, and endotoxins from lipid peroxidation induce inflammation, oxidative stress, and fibrosis, which are involved in the development of NASH, cirrhosis, and HCC [7].
PROGRESSION OF LIVER DISEASE
Development of NAFLD
NAFLD progression is determined by hepatic fat accumulation, inflammation, apoptosis, and liver fibrosis [8]. In a healthy liver, various hepatocytes, including hepatic stellate, Kupffer, and immune cells, regulate hepatocyte homeostasis by inactivating the inflammatory signaling pathways [9]. Hepatocytes are densely packed in a healthy liver [10]; however, normal hepatocyte morphology is altered with increased disruption of cellular homeostasis due to lipid accumulation [11]. In the early stages of NAFLD progression, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated cell death pathways are promoted in steatotic hepatocytes or activated hepatic stellate cells due to natural killer (NK) cell activation [10]. In addition, activated NK cells secrete interferons, which increase M1 phase pro-inflammatory cytokine release from hepatic macrophages [11]. Activated inflammatory signaling pathway promotes the IR signaling pathway and fibrogenesis, consequently activating the transition from NAFL to NASH [12]. Moreover, the number of statocysts increases, and inflammation and fibrosis are activated to induce cell death [13]. Empty spaces are replaced by cellular fibrosis or abnormal hepatocyte-derived cancer cells, but not hepatocytes, thereby promoting HCC development (Fig. 1) [10].
Induction of NAFLD progression in an in vivo model
Dietary model: NASH is mainly caused by the dysregulation of lipid synthesis and hyperglycemia due to obesity [14]. Depending on the experimental purpose, researchers have established various pathogenic animal models via dietary modification, transgenesis, or by injecting chemicals into the animals [14]. A high-fat diet (HFD) is used for establishing NAFLD animal models by increasing the cholesterol and fat content of food [15]. By using an HFD, most pathogenic symptoms, including obesity, T2DM, or liver disease, in NAFLD models were similar to those observed in humans [16]. NAFLD mouse models are mostly established by administering diets rich in fat, cholesterol, and fructose [17]. Depending on the ingredients, HFD can be used to establish animal NAFLD and HCC models in as little as eight weeks [18]. Furthermore, HFD-mediated hepatocyte injury in NAFLD or other metabolic diseases induces typical phenotypic characteristics observed in humans, including weight gain, IR, and increased serum levels of aspartate transaminase and alanine transaminase [19]. Various studies have attempted to establish HCC models using HFD with fructose-mixed water [9]. Animal models of HCC can also be established after 24 weeks using water containing 2.31% fructose, which is a trigger of hyperglycemia [20,21].
Methionine and choline-deficient diet (MCD) is used to establish NAFLD models [22]. In principle, methionine is an amino acid that induces protein and lipid metabolism via DNA and RNA methylation [23]. It functions as a regulator of cellular processes and phospholipid synthesis in hepatocytes and protects against NAFLD progression [23]. Methionine deficiency leads to liver injury, inflammation, and liver fibrosis [24]. In addition, choline deficiency activates macrovascular steatosis in NAFLD models [25]. MCD has been reported to induce hepatic steatosis in a 4–8 weeks dietary model of NAFLD [23]. T helper-17 and -22 cells activate pro-inflammatory cytokine release in the NAFLD model, inducing a cascade of inflammatory signaling pathways, such as the toll-like receptor (TLR)-4-mediated Myd88 or nuclear factor of kappa photopolypeptide enhancer (NF-κB) pathways [26], and activating B cells in an MCD animal model [26]. In addition, MCD inhibits the protein kinase B/mechanistic target of rapamycin kinase (AKT/mTOR)-dependent autophagic cell death and macrophage activation in hepatocytes [27]. MCD model exerts similar effects as the HFD model, such as the induction of hepatic steatosis and liver fibrosis, but it cannot mimic weight gain-mediated T2D and its mediated NAFLD-related signaling pathways [25].
Chemically-induced NAFLD models: Streptozotocin (STZ) induces diabetes by destroying the pancreatic β cells via glucose transporter (GLUT)-2-mediated DNA fragmentation [28]. After inducing cell death with STZ, β-cells do not secrete insulin, resulting in T2DM [28]. STAM model is an NAFLD model in which STZ is injected into 2-day-old mice [28]. Previously, various studies administered STZ to 2-day-old mice without insulin secretion to induce pancreatic β-cell death and dysregulation of insulin-mediated glucose uptake [14]. Four weeks later, STZ-injected mice fed an HFD for up to 8 weeks developed hepatic steatosis [28]. Furthermore, administering HFD over 16–24 weeks induces hepatic steatosis in an HCC model [29]. Consistent progression can be observed in a human HFD-fed NAFLD-HCC model induced by diabetes, which is the main advantage of the STAM model (Table 1) [30].
INFLAMMATORY SIGNALING PATHWAYS UNDER LIVER INJURY
Inflammation and its mediated signaling pathways are important for NAFLD to NASH progression [11]. Through dysregulation of oxidative stress, endoplasmic reticulum (ER) stress, or lipid metabolism, inflammatory signaling pathways increase the secretion of inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, and IL-10 [31]. Accumulation of inflammatory cytokines also induces liver cell apoptosis and fibrosis by activating the transforming growth factor-β signaling pathway [32].
Inflammatory cytokine-mediated NAFLD development
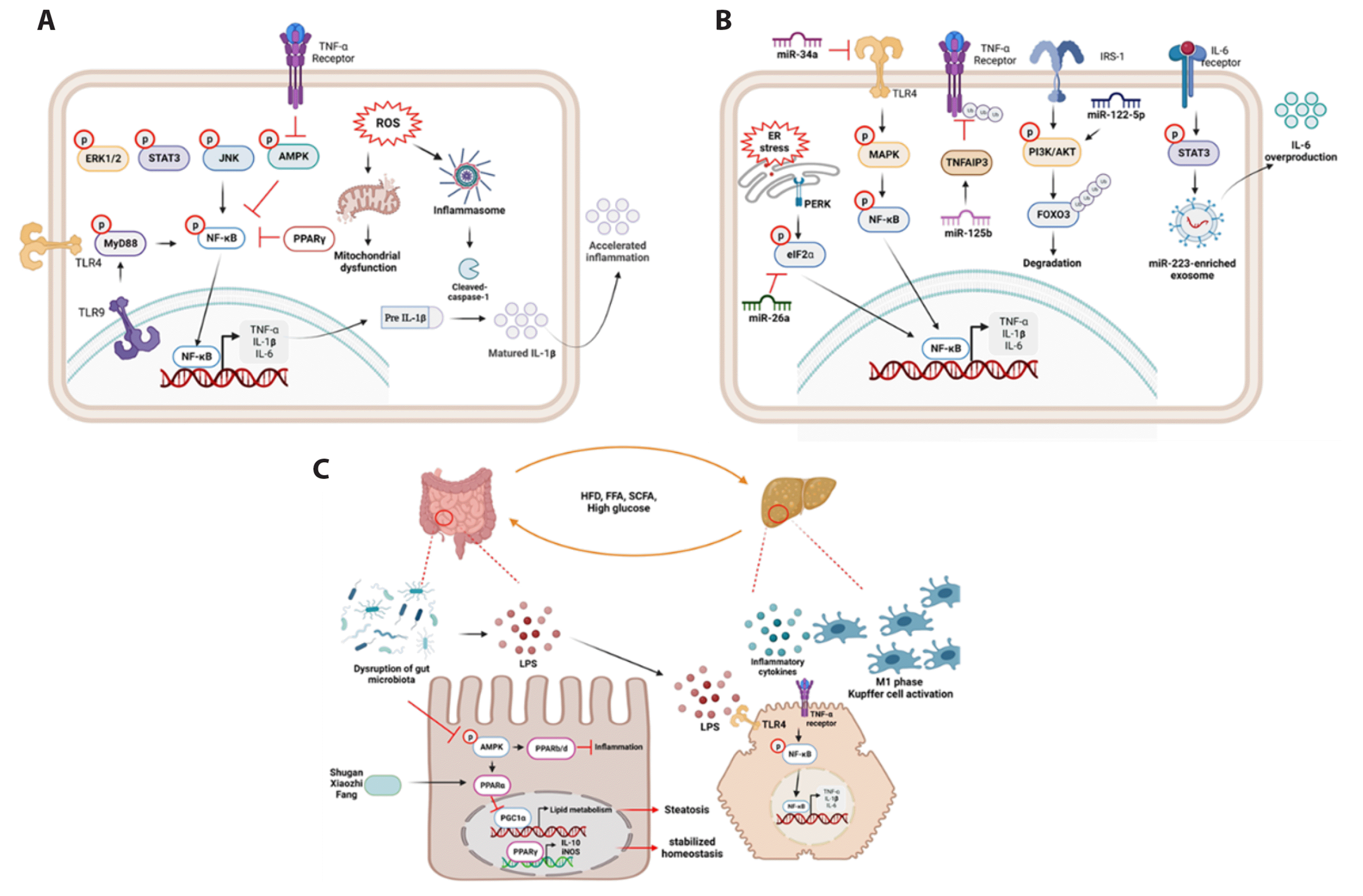
NF-κB activation is a key factor in inflammatory cytokine release [33]. Promoter activation of NF-κB induced by inflammatory cytokines or other mechanisms induces cell apoptosis, inflammation, and fibrosis [34]. HFD or MCD induce NF-κB phosphorylation and TNF-α and IL-1β secretion [35,36]. AMP-activated protein kinase (AMPK) is a sensing marker of energy metabolism that is inactivated by HFD-induced NF-κB activation via peroxisome proliferator-activated receptor (PPAR)−γ promoter activation [37]. In addition, palmitic acid (PA), a free fatty acid (FFA), activates NF-κB phosphorylation-mediated TNF-α, IL-1β, and IL-6 production by suppressing AMPKα expression in hepatocytes [38]. TLR4 is a major receptor inducing Myd88-mediated NF-κB p65 phosphorylation and increasing mRNA expression of TNF-α and IL-1β, thereby causing liver damage [39]. HFD induces TLR4 expression and activates the TLR4/NF-κB signaling pathway to induce liver inflammation [40]. In addition, MCD model induces liver inflammation via TLR4 or 9-mediated Myd88/NF-κB activation, which further induces TNF-α, IL-6, and cleaved caspase-1-mediated IL-1β maturation [35]. NF-κB activation caused by HFD activates mitochondrial dysfunction-mediated reactive oxygen species (ROS) production via dysregulation of the mitochondrial respiratory chain reaction [41]. Mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) signaling pathways act as inflammatory signaling pathways upon inflammatory cytokine stimulation [42]. MAPK consists of extracellular signal-regulated kinase-1/2, p38, and c-Jun N-terminal kinase that activates liver inflammation via production of TNF-α, IL-6, and IL-1β [43]. In addition, high levels of ROS increase damage-associated molecular pattern-mediated NLR family pyrin domain-containing 3 (NLRP3) activation, and pro-inflammatory cytokines released by inflammasome expression significantly activate stellate cell fibrosis [44]. Fibrosis in stellate cells increases STAT3-mediated HCC progression [44]. In addition, damage or pathogen-associated molecular pattern-mediated NLRP3 inflammasome increases cleaved caspase-1 activation and IL-1β maturation in hepatocytes [45]. Mature IL-1β is involved in NLRP3 inflammasome internalization into hepatic stellate cells, and its inflammatory signaling pathway increases collagen production-mediated liver fibrosis (Fig. 2A) [45].
Effect of exosomal activity on NAFLD inflammation
MicroRNAs (miRNAs) consist of 20–25 nucleotides and control target gene expression by suppressing mRNA expression [46]. Dysregulated miRNA expression can control NAFLD inflammation by activating the NF-κB, MAPK, and TLR4 signaling pathways, or dysregulation of cellular homeostasis [47]. Inhibition of miR-125b suppresses FFA-treated NF-κB p65 phosphorylation via direct binding with TNF-α-induced protein 3, which is a negative regulator of NAFLD progression in HepG2 cells and animal models [48]. Increased miR-122-5p expression and secretion of inflammatory cytokines, including TNF-α, IL-6, and IL-8, are observed in HFD- and PA-treated HepG2 cells [49]. Mechanistically, HFD or PA-induced miR-122-5p directly inhibits forkhead box-O3 expression and activates the PI3K/AKT/mTOR signaling pathway [49]. In addition, STAT3 phosphorylation caused by the IL-6 signaling pathway increases miR-223-enriched exosome secretion in patients with NAFLD/NASH [50]. Chronic ER stress activation is also involved in NAFLD pathogenesis [51]. Briefly, in HFD-treated cell chronic ER stress signaling pathway, miR-26a overexpression blocks the phosphorylation of eukaryotic translation initiation factor 2 alpha in a negative feedback loop [51]. miR-34a exerts protective effects on HFD-mediated TNF-α release by suppressing TLR4-induced NF-κB activation [52]. Additionally, miR-34a decreases lipid absorption, lipogenesis, and autophagy by activating the sirtuin 1 (SIRT1)-mediated sterol regulatory element-binding transcription factor 1c (SREBP1c) signaling pathway (Fig. 2B) [52,53].
Effects of gut microbiota on NAFLD progression
Numerous studies have reported that dysbiosis of the gut microbiota caused by HFD or alcohol affects NAFLD pathogenesis [54]. Dysregulation of gut microbiota caused by bacteria, protists, archaea, fungi, and viruses induces the secretion of inflammatory factors, gut hyperpermeability, ethanol, and LPS stimulation [54]. Accordingly, various secreted factors move into the liver through the portal veins and aggravate the inflammatory signaling pathway, progressing NAFLD fibrosis to HCC by activating oxidative stress, ER stress, lipid accumulation, and hepatocyte cell death [55]. In particular, during the progression of NAFLD or HCC, peripheral blood mononuclear cells secrete inflammatory cytokines, such as IL-6 or IL-10, and their inflammatory signaling pathway increases the number of cytotoxic clusters of differentiation (CD)8+ T cells [56]. Moreover, gut barrier permeability during NASH progression, LPS levels were increased by MCD, and LPS induced liver cell inflammation, oxidative stress, and IR by suppressing glucagon-like peptide 1 (GLP1) and 2 expression [57].
PPAR is a group of fatty acid sensors, and previous studies have indicated that PPARs activation suppresses the disruption of gut microbiota-mediated NAFLD progression [58]. PPARs consist of α, β, δ, and γ, and their components show protective effects on NAFLD inflammation through various signaling pathways [58]. First, PPARα activation can prevent HFD-induced NAFLD steatosis in models by activating the AMPK activation-mediated PPARγ coactivator-1α (PGC-1α) signaling pathway [59]. In addition, treatment with the Shugan Xiaozhi decoction, a component of artemisiae scopariae herb, significantly suppressed the gut-liver axis in NAFLD models by activating PPAR-mediated lipid metabolism [60]. Previous studies have indicated that PPARβ/δ shows high metabolic activity, lipid metabolism, glucose homeostasis, and inflammation in hepatocytes, Kupffer cells, and hepatic stellate cells through AMPK activation-mediated suppression of the inflammatory signaling pathway [58]. In addition, short-chain fatty acid-mediated PPARα and γ expression lead to IL-10 secretion and inducible nitric oxide synthase, which stabilizes gut homeostasis and inhibits colonization of pathogenic bacteria [61] (Fig. 2C).
LIVER CELL APOPTOSIS IN NAFLD AND NASH
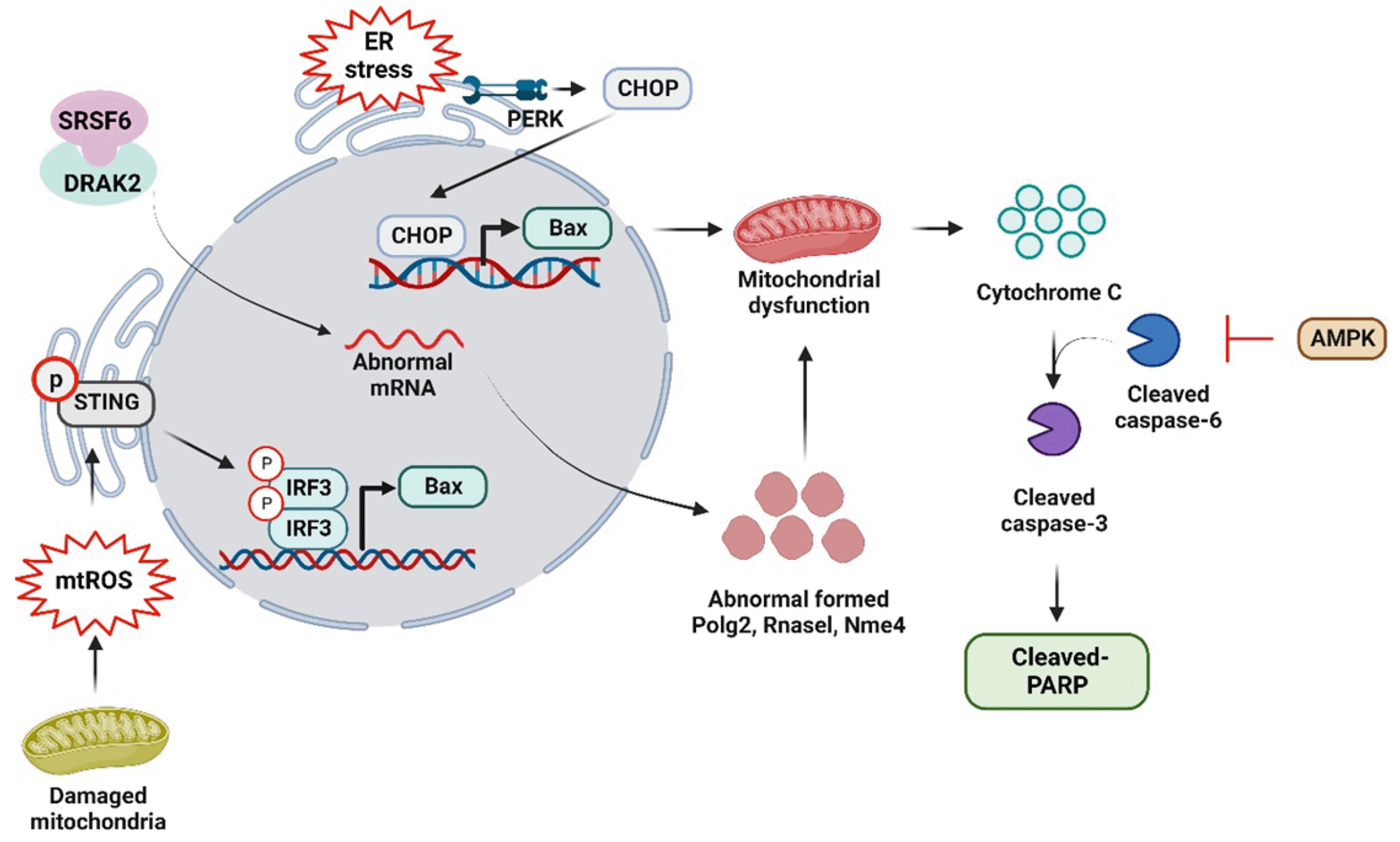
Apoptosis is a marker of NASH to HCC progression [62]. In patients with NASH, it has been found that terminal deoxynucleotidyl transferase dUTP nick end labeling-positive hepatocytes and cleaved caspase-3 expression are higher than in healthy liver specimen [62]. Additionally, overstimulated inflammatory signaling pathway treated with high glucose and HFD-induced TNF-α secretion induced liver cell apoptosis by activating cleaved caspase-3 and -8 [63]. Another apoptosis marker, cleaved caspase-6 activation, suppressed by AMPK activation, inhibited liver cell apoptosis and steatosis [64]. B-cell lymphoma-associated X (Bax) is one of the markers of the mitochondria-mediated apoptotic signaling pathway; when cell apoptosis occurs, B-cell lymphoma expressed on mitochondria membranes is decreased and Bax expression is increased simultaneously [65]. Hyperglycemia and hyperlipidemia-induced by FFA increased dysregulation of glucose and lipid metabolism in the liver and increased apoptosis through stimulator of interferon gene (STING)/interferon regulatory factor 3 (IRF3)-mediated Bax, cleaved-caspase 3, and cleaved PARP activation [66]. In addition, mitochondrial ROS (mtROS) production, which is one of the reasons for mitochondrial dysfunction [67], can increase the protein expression of STING/IRF3 and its mediated apoptosis signaling pathway [41]. In addition, a complex of death-associated protein kinase-related apoptosis-inducing kinase-2 with serine/arginine-rich splicing factor 6 activation in NAFLD models induces mitochondrial dysfunction by inducing RNA splicing-mediated liver apoptosis [68]. Moreover, mitochondrial dysfunction results in excess production of ROS and calcium, which can cause ER stress-mediated cell apoptosis in patients with NAFLD [68]. In particular, Bax expression is regulated by c/EBP homologous protein (CHOP), a marker of ER stress-mediated apoptosis [67]. NAFLD model estimated by HFD-fed ApoE-/- mice for five weeks show significantly increased inflammatory and ER stress pathways, including protein kinase RNA-like ER kinase/activating transcription factor (ATF)-4, inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE)1/x-box binding 1 (XBP-1), and ATF6-mediated CHOP pathways [69]. CHOP expression results in excessive mTOR-dependent autophagy, inflammation, and apoptosis in hepatocytes via activation of the ER stress signaling pathway in HFD-fed ApoE-deficient mice (Fig. 3) [69].
INVOLVEMENT OF LIPID METABOLISM IN NAFLD PROGRESSION
Lipid metabolism plays an important role in NAFLD progression [70]. There are various reasons for the dysfunction of lipid metabolism, such as (1) overproduction of lipid moved to the liver caused by excessive calorie intake, (2) overproduced FFAs in adipose tissue, (3) activated de novo lipogenesis, (4) suppression of triglyceride (TG) content by very low-density lipoprotein, and (5) inactivation of lipid oxidation in the liver [69]. Accumulation of FFAs in the blood can cause IR and is divided into two signaling pathways depending on the phosphorylation site of insulin receptor substrate (IRS)1 [71]. Normally, phosphorylation at tyrosine 895 on IRS activation leads to PI3K/Akt-mediated GLUT4 uptake in skeletal muscle cells [72]. Moreover, high levels of insulin in the blood increase FFA synthesis by activating acetyl-coenzyme A [72]. Accumulation of FFAs increases NAFLD steatosis and inflammation, thereby inducing HCC progression [72].
De novo lipogenesis
Patients with NAFLD exhibit over 2 times higher expression levels of de novo lipogenesis markers than the health controls [73]. Various nuclear receptors or cytoplasmic transcription factors are involved in NAFLD progression by accumulating PPAR, a member of nuclear receptor or SREBP1 and carbohydrate response element (ChREBP), and cytoplasmic transcription factor-mediated de novo lipogenesis [73]. SREBP1 expression is increased by insulin, oxysterol binding protein, or liver X receptor and induced by the IR-mediated Akt signaling pathway [66]. Activation of SREBP1 in de novo lipogenesis stimulates FFA synthesis by stimulating the expression of ACC and FAS [74]. Previous studies have reported that STING/IRF3 activation induces SREBP1-mediated liver cell steatosis and inflammation in HFD-fed NAFLD mouse models [66]. In addition, short-chain fatty acids attenuated MCD-induced SREBP1 and AMPK deactivation-mediated disruption of the antioxidant signaling pathway in hepatocytes [26]. MCD-induced SREBP1 expression increases NF-κB p65 activation and its mediated inflammatory signaling pathway is involved in NASH progression [75]. Suppression of SREBP1 caused by ginsenoside Rg1 treatment significantly increases AMPK expression and suppresses FAS protein expression [38]. Moreover, HFD-mediated SIRT1/AMPK inactivation increases SREBP1-mediated lipogenesis, PGC-1α-induced mitochondrial dysfunction, oxidative stress, and NF-κB-mediated inflammation [36]. A previous report revealed that HFD with high fructose induced TLR4-mediated TNF-α stimulation [76]. At the same time, the de novo lipogenesis pathway was turned on, which increased the protein expression of SREBP1 and FAS [76]. Along with SREBP1, ChREBP-1 also induces de novo lipogenesis and functions as a cytoplasmic transcription factor by inducing pyruvate kinase expression and TG synthesis [73]. High-fructose diet increases ChREBP-1-associated hepatic steatosis by activating de novo lipogenesis and LPS production [77]. Silencing of NF-κB p65 expression using siRNA transfection suppresses IR-mediated ChREBP-1 nuclear translocation, indicating that excessive liver inflammation affects liver steatosis [78]. HFD-mediated increase in ChREBP-1 and SREBP1 expression is suppressed by small heterodimer partner interacting leucine zipper protein, nuclear receptor activation, which is increased by lyophilized maqui berry extract treatment [79].
Lipid oxidation
Lipid oxidation is essential for regulating mitochondrial biogenesis and cellular homeostasis in liver cells [80]. In a healthy liver, mitochondria use FFAs as an energy source [80]. Mechanistically, when FFAs enter the mitochondria, they are converted by acetyl CoA and then conjugated with carnitine, which is located on the outer membrane of the mitochondria [80]. Subsequently, the acetyl CoA-carnitine form was disassociated by carnitine palmitoyl transferase (CPT)-2, and acetyl CoA was degraded by β-oxidation [80]. However, increased influx of FFAs induced mutation of mitochondrial DNA, mitochondrial dysfunction, and increased ROS-mediated inflammatory signaling pathway in hepatocytes [81]. AMPK is a protein kinase that switches lipid synthesis by activating sucrose non-fermenting-1 expression [81]. Based on the function of AMPK in lipid oxidation, PPARα activation is related to β-oxidation by inducing FFA in the mitochondria and CPT-1 activation [82]. Inactivation of AMPK phosphorylation by compound C, an inhibitor of AMPK activation, increases liver cell steatosis by increasing FAS mRNA expression, and HFD-mediated AMPK inactivation causes SIRT1 activation [81]. AMPK-mediated SIRT1 expression suppresses HFD-mediated lipid oxidation by suppressing PGC1-α, SREBP1, SCD-1, and FAS and increasing PPARα expression levels [36]. AMPK phosphorylation increases PPARα-associated fatty acid oxidation and suppresses NF-κB-mediated inflammatory cytokine release [37]. Additionally, the SIRT1 and PGC1α axis activated by leptin or adiponectin-mediated AMPK activation stabilized mitochondrial function and increased fatty acid oxidation in hepatocytes [83]. Moreover, methylation of the controlled J protein, a protein on mitochondria respiratory chain complex 1, is highly expressed in patients with NAFLD, and its overexpression leads to lipid accumulation, suppression β of-oxidation, and hepatocyte damage [84].
GLUCONEOGENESIS
Hepatic gluconeogenesis refers to the production of glucose by the GLUT2 signaling pathway under fasting conditions, primarily in response to insulin and glucagon levels [85]. Activation of pyruvate to phosphoenolpyruvate is the first step for phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphate (G6Pase)-mediated gluconeogenesis [85]. Excessive release of glucose via gluconeogenesis enhances IR-mediated inflammation, steatosis, and de novo lipogenesis in patients with NAFLD [86]. PGC-1α-activated PEPCK expression induced by HFD-mediated inflammation is suppressed by bicyclol, an approved drug for targeting hepatitis, liver fibrosis, and liver injury [86]. Inactivation of the AMPK-mediated SIRT–PGC1 axis is involved in HFD-fed NASH progression [83]. PEPCK and G6Pase activation are modulated by PGC-1α activation and produce glucose in T2DM models [87]. Upstream of PEPCK-mediated gluconeogenesis, glucagon or FFA activates the formation of CREB-regulated transcription coactivator (CRTC)-2 with CEBP, and its complex increases PEPCK gene expression as a transcription factor [86]. HFD-mediated salt-induced kinase 1 suppression leads to CRTC2-mediated PEPCK and G6Pase gene expression and increases hyperglycemia and liver steatosis [88]. ER stress-mediated inhibition of AMPK phosphorylation increases the phosphorylation of CREB and PEPCK proteins [89].
ANTI-FIBROTIC DRUGS IN NAFLD/NASH TREATMENT
NASH is a liver disease characterized by the buildup of fat in the liver, which causes inflammation and damage to liver cells, leading to fibrosis (scarring) and cirrhosis [90]. Several drugs are being developed for the treatment of NASH by exerting anti-fibrotic effects on the liver [91]. Liver fibrosis occurs when there is chronic damage to the liver, such as inflammation and oxidative stress, which leads to the accumulation of scar tissue. This scar tissue can impair liver function and, if left unchecked, progress to cirrhosis, liver failure, and HCC [90]. Therefore, anti-fibrotic drugs can help slow or even reverse liver fibrosis by inhibiting the production of collagen fibers, promoting the breakdown of scar tissue, and reducing inflammation in the liver [91]. These drugs can potentially reduce the risk of liver-related complications and improve the overall prognosis of patients with NASH. Several different classes of drugs have been studied for their anti-fibrotic effects in NASH, including peroxisome PPAR agonists, farnesoid X receptor (FXR) agonists, GLP-1 receptor agonists, and C–C chemokine receptor (CCR) 2/5 inhibitors [92,93]. PPAR agonists, including pioglitazone and elafibranor, regulate lipid metabolism and reduce inflammation in the liver [92]. Recent study suggested that anti-diabetic drugs with a potential anti-inflammatory effect can ameliorate the manifestations of NAFLD, and thus it can be a therapeutic option for such a condition came from metabolic syndrome [92]. In addition to their anti-fibrotic effects, many NASH drugs have other beneficial effects on the liver, such as reducing hepatic steatosis (fat accumulation), improving insulin sensitivity, and reducing inflammation [94]. For example, some drugs that target FXR can also improve lipid metabolism and reduce hepatic inflammation, in addition to their anti-fibrotic effects [94]. Furthermore, in clinical trials, CCR2/5 inhibitors have shown promise in improving liver fibrosis and inflammation in patients with NASH [95]. In another study, patients with NASH receiving maraviroc, another CCR2/5 inhibitor, exhibited a significant reduction in liver inflammation compared to those receiving placebo [96]. Therefore, anti-fibrotic drugs are important components of the multidisciplinary approach to treat NASH. Further research is needed to identify new and effective therapies for treating NAFLD/NASH.
NEW STRATEGIES FOR THE DEVELOPMENT OF DRUGS FOR NAFLD AND FUTURE DIRECTIONS
Accumulating evidence indicates that GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) agonists can improve NAFLD. These drugs have been shown to reduce liver fat content, improve liver enzymes, and decrease markers of inflammation in patients with NAFLD [97-99]. Various GLP-1 agonists, such as liraglutide, semaglutide, and exenatide, have significantly reduced the liver fat content and improved the liver enzymes in clinical trials [100]. GIP agonists have also shown promising results in reducing liver fat content and improving insulin sensitivity [100]. In addition to their effects on liver fat, GLP-1 and GIP agonists have been shown to improve cardiovascular risk factors, such as blood pressure, lipid levels, and markers of inflammation [98,101]. These drugs may also have a protective effect on the pancreas, reducing the risk of pancreatic fat accumulation and pancreatitis [98,101]. Tirzepatide is a novel dual GIP and GLP-1 receptor agonist [98]. It is currently being evaluated in clinical trials as a potential therapeutic for type 2 diabetes and obesity [98]. There is limited data available on the effects of tirzepatide on NAFLD, as most clinical trials have focused on its effects on glycemic control and weight loss [98,102]. However, some studies have investigated the potential benefits of GLP-1 and GIP agonists, including tirzepatide, in NAFLD [97,98]. One randomized control trial evaluated the effects of semaglutide, a GLP-1 agonist, in patients with NASH, a more severe form of NAFLD [103]. The trial found that semaglutide significantly improves liver histology, with 59% of patients showing improvement in fibrosis and 43% showing resolution of NASH [103]. Overall, GLP-1 and GIP agonists appear to be promising therapeutic agents for NAFLD treatment. However, further studies are needed to determine the optimal dosing and duration of treatment as well as the long-term safety and efficacy of these drugs in patients with NAFLD.
Despite several efforts to develop treatments for NAFLD, the unmet need for effective drug therapies remains significant. The complexity of the disease, lack of understanding of its pathogenesis, and limited availability of relevant preclinical models increase the difficulty of drug development for NAFLD. One potential strategy is to focus on the identification of NAFLD-specific novel therapeutic targets via a comprehensive understanding of the underlying molecular mechanisms and identification of key pathways dysregulated in NAFLD. Another approach is to develop preclinical models that accurately recapitulate the features of human NAFLD. Development of such models will enable more accurate evaluation of the efficacy and safety of potential NAFLD therapeutics. For example, humanized mouse models, in which human liver cells are engrafted into immunodeficient mice, or 3D liver organoids, for study of liver function and disease in vitro, can be used for future studies.
DISCUSSION
In this review, we have outlined the inflammation, apoptosis, lipid metabolism, and gluconeogenesis mechanisms involved in NAFLD progression. Various pathways, such as PPAR, NF-κB, MAPK, STAT, and AMPK signaling pathways, are associated with and accelerate the development of liver steatosis and fibrosis. Several studies suggest targeting the PPAR signaling pathway to treat NAFLD progression. PPARs act as transcriptional targets and are highly associated with inflammation, liver steatosis, IR, and liver fibrosis. Diverse natural products exert pharmacological effects in suppressing liver fibrosis, ROS production, and lipid metabolism. However, the detailed molecular mechanisms and functions of organelles involved in NAFLD progression remain unknown and require further investigation. Additionally, natural products should be modified with effective drugs by targeting specific molecules to prevent NAFLD progression. In summary, determination of the underlying mechanisms and modification of natural compounds for drug development are potential strategies for the treatment of NAFLD and other metabolic diseases, such as T2DM and cardiovascular disease.
 XML Download
XML Download