

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
As people’s lifestyle has been changing, the incidence rate of hypertension rises year by year. Statistics suggests that nearly 50% of deaths from cardiovascular disease are associated with hypertension. Myocardial injury caused by pressure overload that is accompanied by hypertension shows a close association with cardiovascular death. Cardiomyocyte hypertrophy and apoptosis may occur under the pressure overload, and myocardium is gradually replaced by fibrosis tissues (e.g., type I and type III fibrous collagen, elastin, fibronectin, as well as proteoglycan) [1]. Myocardial fibrosis (MF) acts as a vital link during the myocardial remodeling process in hypertension; it is also reported as the major cause of cardiac failure. Nevertheless, the specific regulatory mechanism of MF under the pressure overload remains unclear. Existing studies have shown that pyroptosis mediates the formation of “pyroptosis-inflammatory response-fibrosis” axis-like pathological changes in lung, liver, kidney and other tissue fibrotic diseases, expands the inflammatory cascade, and aggravates the course of tissue fibrosis [2-4]. Recent studies have also shown that cardiomyocyte pyroptosis is closely bound up with the occurrence of cardiovascular diseases, especially MF [5]. Therefore, elaborating the mechanisms regulating cell pyroptosis and MF will open a new target for the treatment of hypertensive cardiomyopathy.
Autophagy refers to a physiological process, through which sirtuin is degraded, and excessive or injured organelles are eliminated following the lysosomal degradation. Some scholars have demonstrated that autophagy is likely to be activated in myocardial or hypoxic-ischemic injury, and moderate autophagy refers to a process of self-salvation that helps cells adapt to inflammation, anoxia, reperfusion injury, as well as other stress responses [6,7]. Nevertheless, through excessive autophagy, cells will be damaged, so the apoptosis or death of cells will be promoted [8]. Some existing studies have suggested that phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway is another classic signaling pathway related to the regulation of autophagy [9]. And the previous research of the research group also showed that activating the PI3K/AKT1 signaling pathway can improve MF [10]. Nevertheless, MF under autophagy-induced stress overload and the internal regulatory mechanism remain incompletely studied. Recently, Li et al. [11] have shown that DHA can inhibit pyroptosis by activating the PI3K/AKT1 pathway, which in turn alleviates ischemia-reperfusion injury. Liu and Tie [12] have also shown that the new target for the treatment of atherosclerosis may be closely bound up with the activation of the PI3K/AKT1 pathway to inhibit pyroptosis. Studies have shown that one key site of translational control is the phosphorylation of eukaryotic translation initiation factor 2α (eif2α), which is triggered by four stress-response kinases, and therefore the expression of eif2α phosphorylation tends to increase during disease [13]. And it is gratifying that related studies have shown that eif2α phosphorylation is involved in PI3K signaling [14,15]. Therefore, we speculated whether the improvement of abdominal aorta coarctation (AAC)-induced MF by hydrogen sulfide (H2S) was related to the activation of PI3K/AKT1 signaling pathway by eif2α phosphorylation to inhibitpyroptosis.
H2S, a novel gaseous signaling pathway, has been shown to have intensive biological effects on the body. Besides, it can dilate blood vessels as well as control autophagy, anti-inflammation and anti-apoptosis [16]. However, whether the gaseous signaling molecule H2S can enhance pressure overload-induced MF and its signal transduction mechanism has been rarely discussed. Studies have found that the model of rats with AAC can simulate MF caused by hypertension under the pressure overload. Therefore, this study intends to establish a rat model of MF induced by abdominal aortic constriction and a model of H9c2 cardiomyocyte injury induced by angiotensin II (Ang II, 1 μM) [17], observe the effect of H2S on pressure overload-induced MF in rats, and discuss the effect of eif2α phosphorylation on PI3K/AKT1 signaling pathway.
Go to : 
METHODS
Chemicals and reagents
Sigma-Aldrich provided sodium hydrogen sulfide (NaHS) (207683-19-0). Wuhan Boster Biological Technology, Ltd. Proteinch provided the antibodies for ATG16L1 (19812-1-AP), Beclin1 (11306-1-AP), MMP1 (10371-2-AP), MMP13 (18165-1-AP), TIMP1 (16644-1-AP), IL-6 (21865-1-AP), ICAM-1 (60299-1-Ig), Caspase1 (22915-1-AP), Caspase3 (19677-1-AP), CTH (12217-1-AP). Cell Signaling Technology provided Rabbit polyclonal P-eif2α (Rabbit mAb#9722), anti-P-PI3K (Rabbit mAb#17366), anti-P-AKT1 (Rabbit mAb#9018), polyclonal anti-GAPDH (Danfoss, Rabbit mAb #5174). Abcam provide IL-1β (ab254360), IL-18 (ab191860), Cleaved-Caspase3 (ab52072), Beyotime Institute of Biotechnology provided phenyl methyl sulfonyl fluoride (PMSF; PB0425), Sigma-Aldrich provided Bicinchoninic Acid (BCA) Protein Assay kit (B9643), and Beyotime Institute of Biotechnology provided Enhanced Chemiluminescence Reagent kit (P0018S), SDS-PAGE Gel Preparation kit and PMSF (P0690).
Establishment of model
We maintained 44 male Sprague–Dawley (SD) rats (slacker company) weighing 220 ± 30 g in a constant humidity environment at a steady temperature (24°C ± 3°C), under a 12 h-day-night cycle and free access to water and feed, 44 experimental rats were split into 4 groups in a random manner: normal rats administrated with H2S group (H2S group), AAC and administrated with H2S group (AAC + H2S group), AAC-treated group (AAC group), normal group (control group). Following skin preparation, the rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (350 mg/kg; The rats showed no signs of peritonitis, pain or discomfort). In the AAC + H2S group and the AAC group, exposing the abdominal aorta above the renal artery, carefully separate the abdominal aorta, fix a blunt needle with a diameter of 7 mm around the abdominal aorta above the renal artery, then ligate 5 mm above both renal arteries and quickly withdraw the blunt needle, resulting in lumen stenosis. During the whole procedure, the rectal temperature was maintained at 36.5°C–37.5°C with a heating lamp. Rats in the control group and H2S group underwent the same procedure, whereas the abdominal aorta was not narrowed. All groups were administrated with penicillin 100,000 units/animal for anti-infection for 3 days once a week. AAC + H2S group and H2S group underwent intraperitoneal injection of NaHS (100 μmol/kg). The rats in the control group and AAC group were daily administrated with PBS. After 4 weeks, they were weighed and anesthetized with chloral hydrate (350 mg/kg, intraperitoneal anesthesia), the cervical spine was dislocated, and the heart was removed and weighed, and then incubated at –80°C for experiments and all the experiments were approved by the University Committee on the Use and Care of Animals of South China University (SYXK2020-0002).
H2S content assay
Myocardial H2S content was assayed by ELISA kit (Beyotime Institute of Biotechnology). Then add 50 µl of diluted (1:1) standard to the reaction well, add 50 µl of the test sample to the reaction well, and incubate at 37°C for 1 h. Remove the liquid in the hole and wash three times. Add 80 µl of affinity streptin-HRP to each well, gently shake and mix, incubate at 37°C for 30 min, and wash three times. Add 50 µl each of substrate A and B to each well, shake gently to mix, and incubate at 37°C for 10 min. Finally, add 50 µl of stop solution quickly, and measure the result immediately after adding the stop solution.
Masson’s staining
Take the left ventricle of rats was fixed in 10% neutral formalin, paraffin embedded, sections were achieved 4 µm, rigorously following the instructions Masson staining steps. Subsequently, different fields of view at 40 × 10 magnification were selected, sections were placed under a microscope for observation.
Transmission electron microscope (TEM) observation
Four weeks later, the rats were killed in a humanitarian manner, left ventricular apex rapidly small piece of tissue was taken and placed in 2.5% glutaraldehyde at 4°C fixed 3 h, remove the glutaraldehyde solution was immersed in PBS at 4°C fixed line TEM inspection.
Western blotting assay
After taking each group of grinding myocardium cell lysate and PMSF, harvested supernatant was quantified in a colorimetrical manner BCA. After being extracted, we heated the protein to 95°C (10 min) and then underwent denaturing and glue, electrophoresis processes. Subsequently, it was placed to a membrane, a hatched anti, Tris buffered saline Tween rinsed, incubated secondary antibody (goat anti-rabbit, goat anti-mouse), and lastly obtained by ECL (Beyotime, Institute of Biotechnology). Using the Alpha Imager software (Alpha Imager Software) for each target protein bands for gray scale scanning, respectively, in comparison with GAPDH gradation value, after statistical analysis using GraphPad Prism 5 (GraphPad Software).
Cell culture
With obtaining the H9c2 cell line from ATCC, the cells were cultured in standard DMEM supplemented with 10% calf serum (Gibco), and during the experiment, the CO2 concentration and air concentration in the incubator were ensured to be 5% and 95%, respectively, and the temperature was humidified at 37°C. Cells were randomly seeded onto 6-well dishes when them grew to a certain density, and treated them with NaHS (400 μM; H2S dononer) for 30 min before H9c2 cardiomyocyte injury was induced using Ang II (1 μM) [17], and the cell lysate was collected for analysis after 24 h.
Statistical analysis
At least three independent experiments were repeated (n = 3). All values are expressed as the mean ± standard error. Student’s t-test was performed with GraphPad Prism software to evaluate statistical significance and one-way analysis of variance (ANOVA) was used to analyze comparisons among multiple groups. But when there are two variables, we use two-way ANOVA. When p < 0.05, it is considered to be statistically significant. The data are statistically evaluated by variance analysis, and then Prism is used for Tukey post-test of inter-group comparison.
Go to :
RESULTS
Variations in rat bodyweight and heart weight/bodyweight ratio
Four weeks later, 10, 10, 9, and 9 rats survived in the control group, AAC group, AAC + H2S group, and H2S group, respectively. Table 1 suggests that the mentioned groups showed no statistically significant differences in bodyweight, heart weight and heart weight/bodyweight ratio (Table 1).
Table 1
Effects of H2S on body weight, heart weight and heart weight/body weight in rats
![]()
Results of rat blood pressure and heart color Doppler ultrasound examination
The cardiac functions of rats were examined by heart color Doppler ultrasound examination, while the arterial blood pressure was ascertained by common carotid artery intubation for each group. According to the results suggested, AAC group and AAC + H2S group left ventricular ejection fraction and fractional shortening not statistically significantly different from those of the control group (p > 0.05). The arterial blood pressures of AAC group and AAC + H2S group were elevated noticeably compared with those of the control group (p < 0.05), and after receiving H2S intervention, AAC + H2S group and AAC group were not statistically significantly different in arterial blood pressure (p > 0.05). The above findings indicate that the AAC rat model under pressure overload was successfully established, while the successfully established model showed no significant loss of cardiac function during the observation period and no anti-hypertensive effect of the H2S donor on the AAC rats (Table 2).
Table 2
Effects of H2S on urine protein, blood pressure, left ventricular ejection fraction and fractional shortening in rats
| Groups | Urine protein (mg/L) | Blood pressure (mmHg) | Left ventricular ejection fraction (%) | Fractional shortening (%) |
|---|---|---|---|---|
| Control | 7.72 ± 1.16 | 84.74 ± 7.88 | 76.66 ± 5.75 | 45.71 ± 6.39 |
| AAC | 6.86 ± 0.77 | 162.70 ± 11.35* | 71.05 ± 6.27 | 38.78 ± 7.30 |
| AAC + H2S | 9.27 ± 0.54 | 155.31 ± 10.02* | 79.94 ± 5.33 | 42.58 ± 3.95 |
| H2S | 10.02 ± 2.16 | 95.19 ± 9.86 | 81.42 ± 7.14 | 41.06 ± 5.77 |
![]()
Change in the level of myocardial endogenous H2S in rats
To explore the in the level variation of myocardial endogenous H2S in rats of each group, the H2S level in myocardial tissue in rats of all groups was ascertained by ELISA. The results showed that compared with the results of the control group, the H2S level in myocardial tissue in AAC group was dropped apparently (p < 0.05), while that in myocardial tissue in AAC + H2S group rose after exogenous H2S donor was added, which showed a statistically significant difference (p < 0.05); H2S group and the control group were not statistically significantly different in H2S level in myocardial tissue (p > 0.05) (Fig. 1).
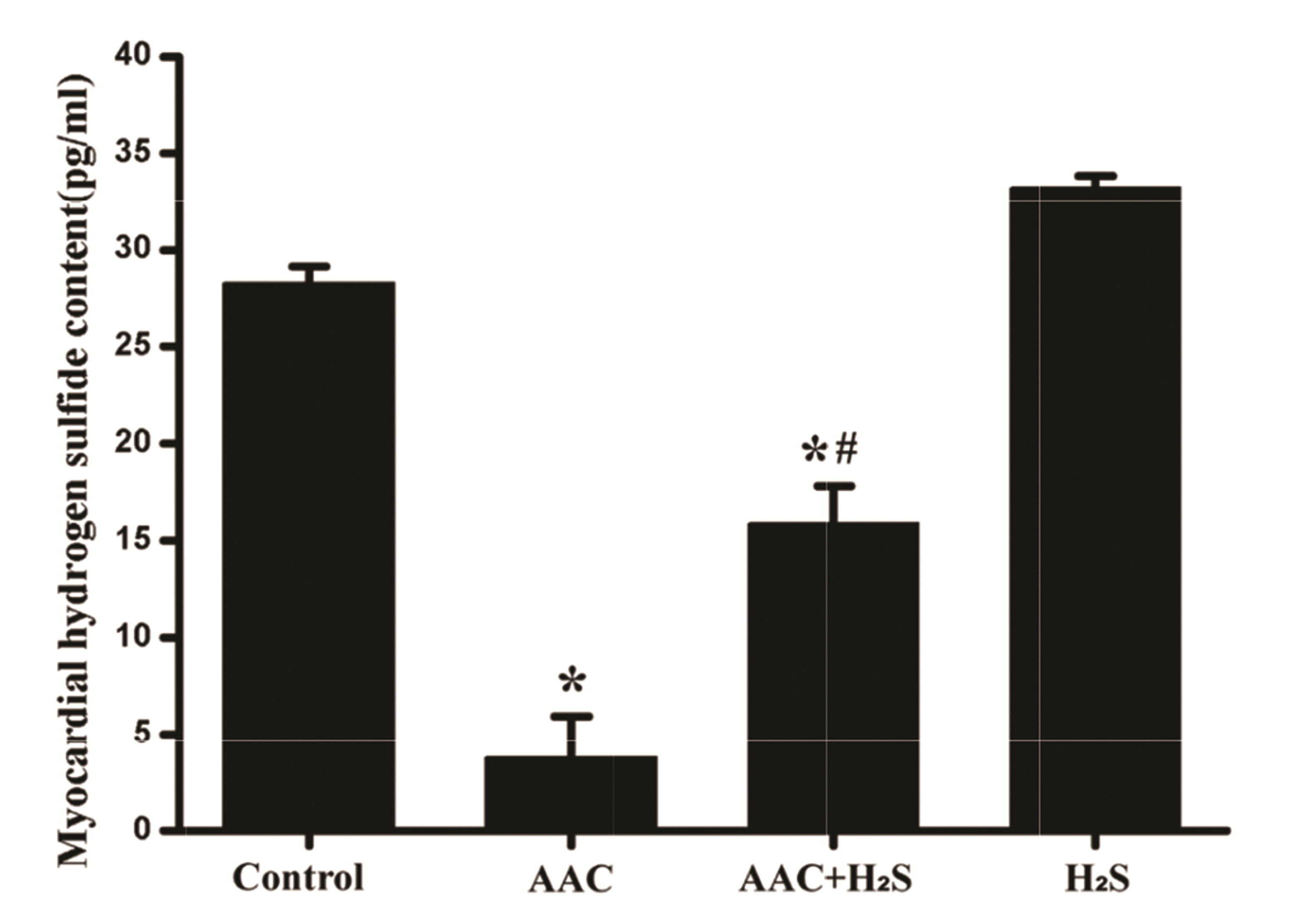
The effect of H2S on pressure overload-induced MF
The deposition of cardiac interstitial collagen and interstitial fibrosis was identified by Masson staining. The Fig. 2 suggests that collagen fibers in AAC group were blue as observed under a microscope, myofilaments were arranged in an irregular manner, and collagen fibers increased more obviously than those of the control group; the deposition of collagen in AAC + H2S group decreased evidently in amount compared with that in the AAC group; the deposition of collagen in H2S group remained nearly unchanged in comparison with the results of the control group (Fig. 2).
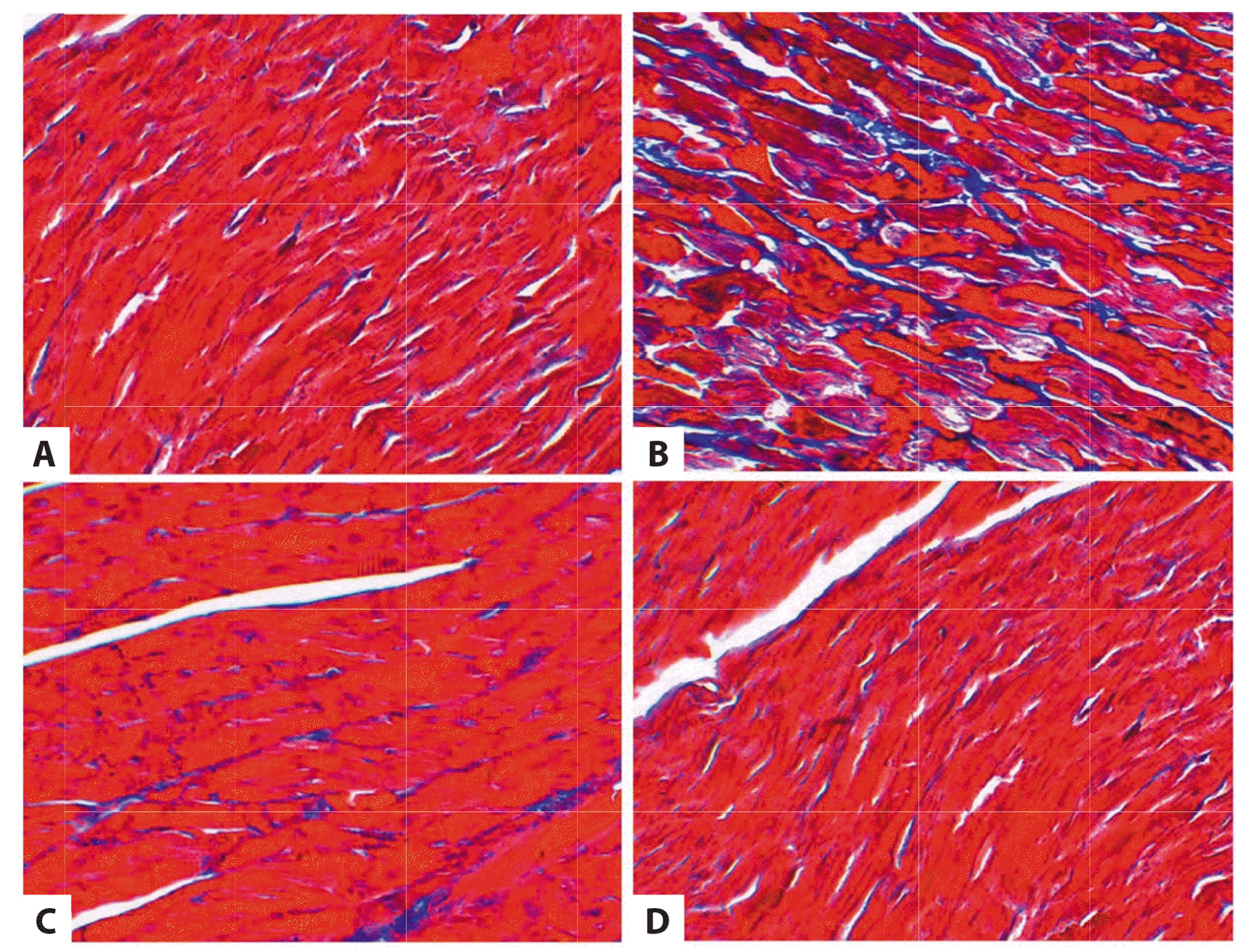 | Fig. 2Upregulation of cardiac production of myocardial collagen fibers in abdominal aorta coarctation (AAC)-injected rat in vivo, this effect can be antagonized by hydrogen sulfide (H2S).Representative images of Masson with myocardial collagen fibers in the heart tissues of AAC-injected rat models (Blue represents collagen fibers). Images were acquired at 10 × 40 magnification. (A) Control group. (B) AAC group. (C) AAC + H2S group. (D) H2S group.
|
The effect of H2S on MMPs/TIMPs expression
Since MMPs/TIMPs dysregulation displays a close association with the excessive deposition of collagen in myocardial interstitium, the variation of the expression of MMPs/TIMPs was detected in this study by Western blotting assay. The result of Western blotting assay suggested that the expression levels of MMP1 (p = 0.0031), MMP13 (p = 0.0368) and TIMP1 (p = 0.0352) in AAC group were significantly up-regulated compared with those of the control group, whereas those of MMP1 (p = 0.0237), MMP13 (p = 0.0286) and TIMP1 (p = 0.0383) in myocardial tissue in AAC + H2S group declined significantly compared with those of the AAC group (p < 0.05), there were no statistically significant differences in the expression levels of MMP1 (p = 0.4081), MMP13 (p = 0.0517) and TIMP1 (p = 0.1826) between H2S group and the control group (Fig. 3).
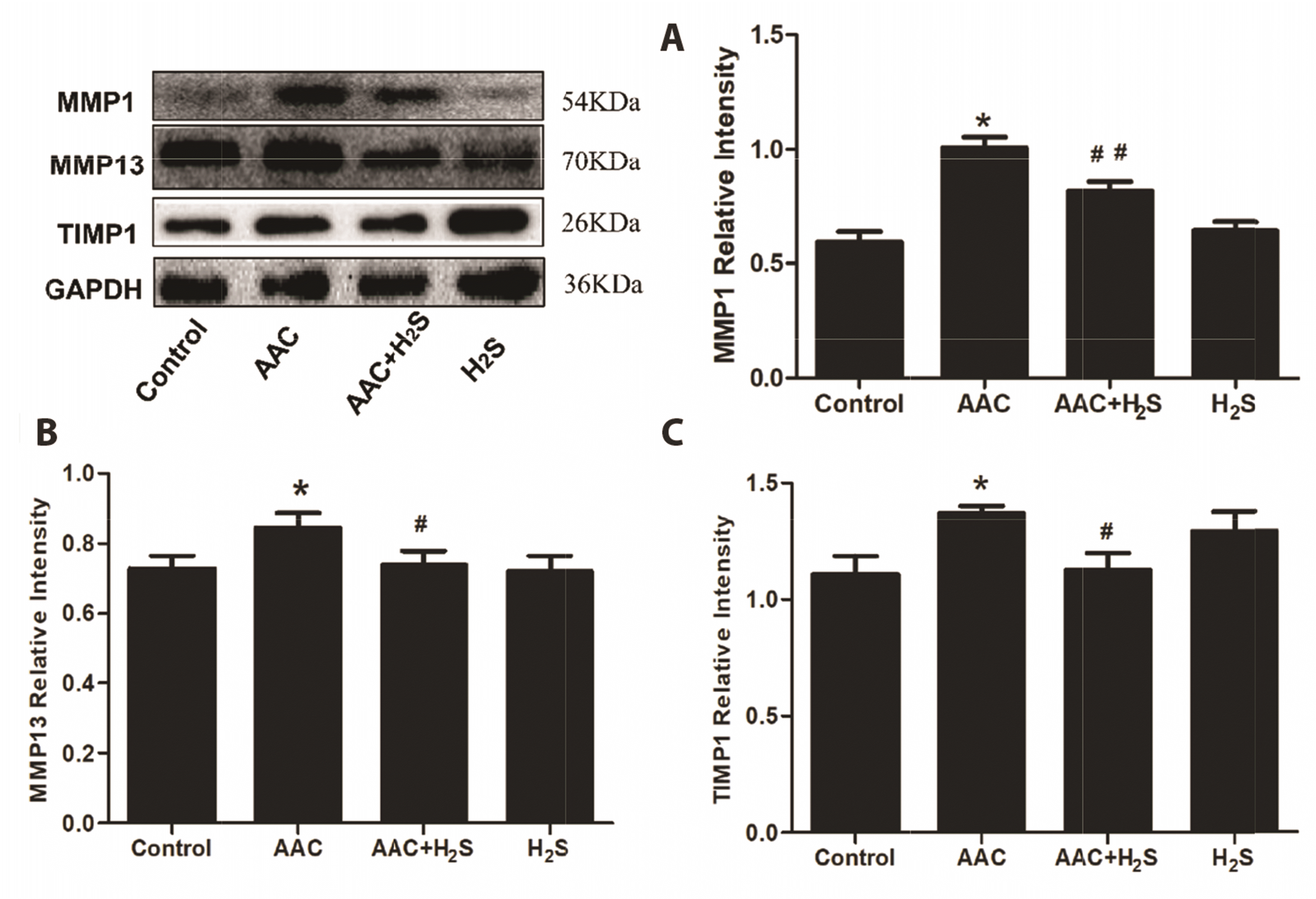 | Fig. 3Western blotting analysis the expression of (A) MMP1, (B) MMP13, (C) TIMP1 in abdominal aorta coarctation (AAC)-induced myocardial fibrosis treated with control, AAC, AAC + H2S, and H2S.Values are presented as mean ± SD. H2S, hydrogen sulfide. *p < 0.05 vs. control, #p < 0.05 vs. AAC, ##p < 0.01 vs. AAC.
|
Effect of H2S on the expression of myocardial pyroptosis-related protein in pressure-overloaded rats
To observe the effect of H2S on pyroptosis in the myocardial tissue of rats under the pressure overload, the expression levels of relevant proteins Caspase1, Caspase3 were detected by Western blotting assay. Our findings revealed that compared with the results of the control group, the protein expression levels of Caspase1 (p = 0.0211), Caspase3 (p = 0.0085), in myocardial tissue in AAC group rose noticeably (p < 0.05), while those of Caspase1 (p = 0.0017), Caspase3 (p = 0.0086) in myocardial tissue in AAC + H2S group declined significantly compared with those of the AAC group. In comparison with the results of the control group, the protein expression levels of Caspase1 (p = 0.9599), Caspase3 (p = 0.9043), in H2S group were almost unchanged, suggesting no differences with statistical significance (Fig. 4).
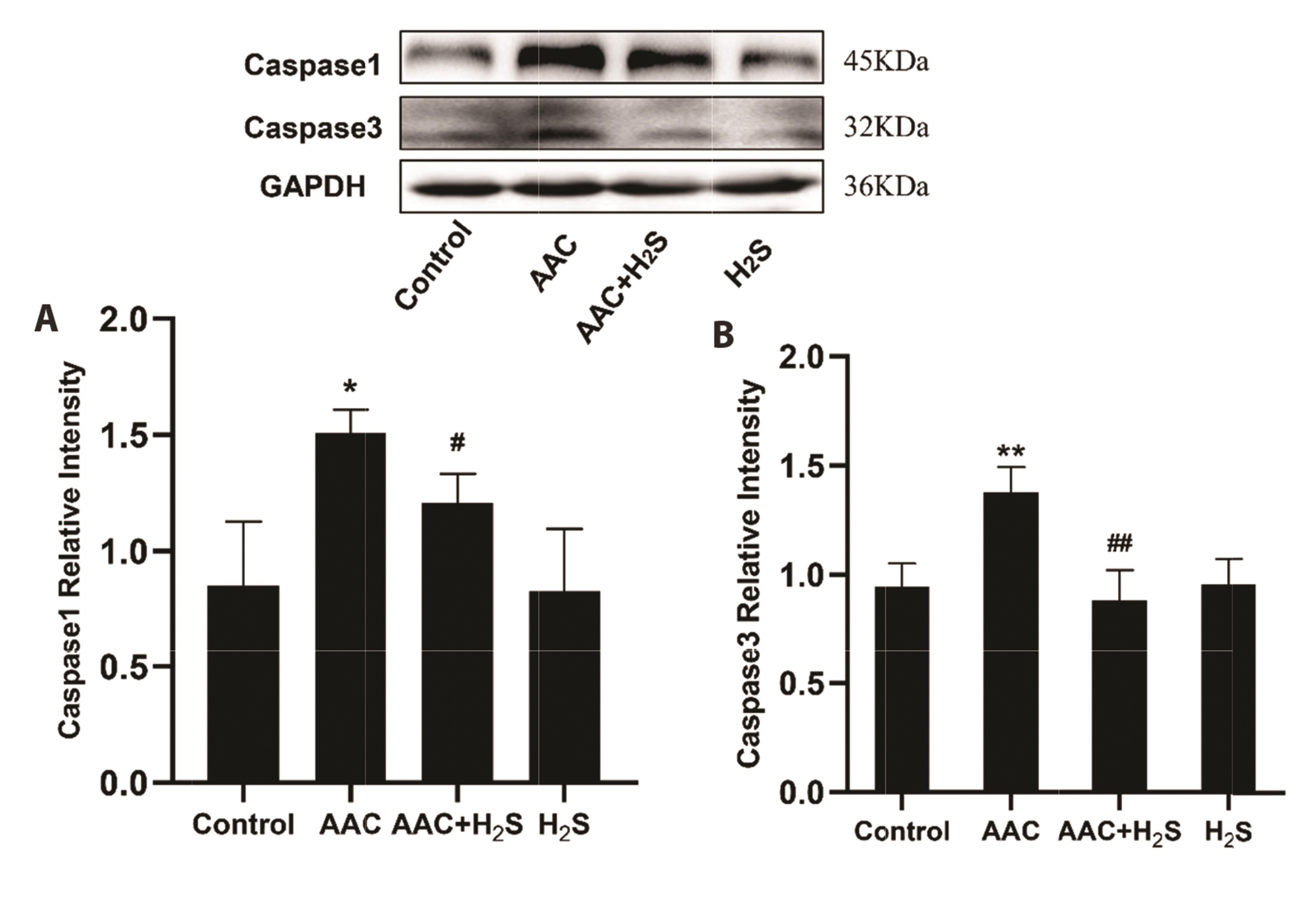 | Fig. 4Western blotting analysis the expression of (A) Caspase1, (B) Caspase3 in abdominal aorta coarctation (AAC)-induced myocardial fibrosis treated with control, AAC, AAC + H2S, and H2S.Values are presented as mean ± SD. H2S, hydrogen sulfide. *p < 0.05 vs. control, **p<0.01 vs. control, ##p<0.01 vs. AAC.
|
The effect of H2S on the expressions of proteins related to myocardial inflammation in rats under the pressure overload
To observe the effect of H2S on inflammation in the myocardial tissue of rats under the pressure overload, the expression levels of relevant proteins IL-6, IL1β, IL18, and ICAM-1 were detected by Western blotting assay. Our findings revealed that compared with the results of the control group, the protein expression levels of IL-6 (p = 0.0158), IL1β (p < 0.0001), IL18 (p = 0.0002) and ICAM-1 (p = 0.0407) in myocardial tissue in AAC group rose noticeably, while those of IL-6 (p = 0.0174), IL1β (p = 0.0221), IL18 (p = 0.0004) and ICAM-1 (p = 0.0088) in myocardial tissue in AAC + H2S group declined significantly compared with those of the AAC group (p < 0.05); In comparison with the results of the control group, the protein expression levels of IL-6 (p = 0.0820), IL1β (p = 0.0614), IL18 (p = 0.0990) and ICAM-1 (p = 0.0635) in H2S group were almost unchanged, suggesting no differences with statistical significance (Fig. 5).
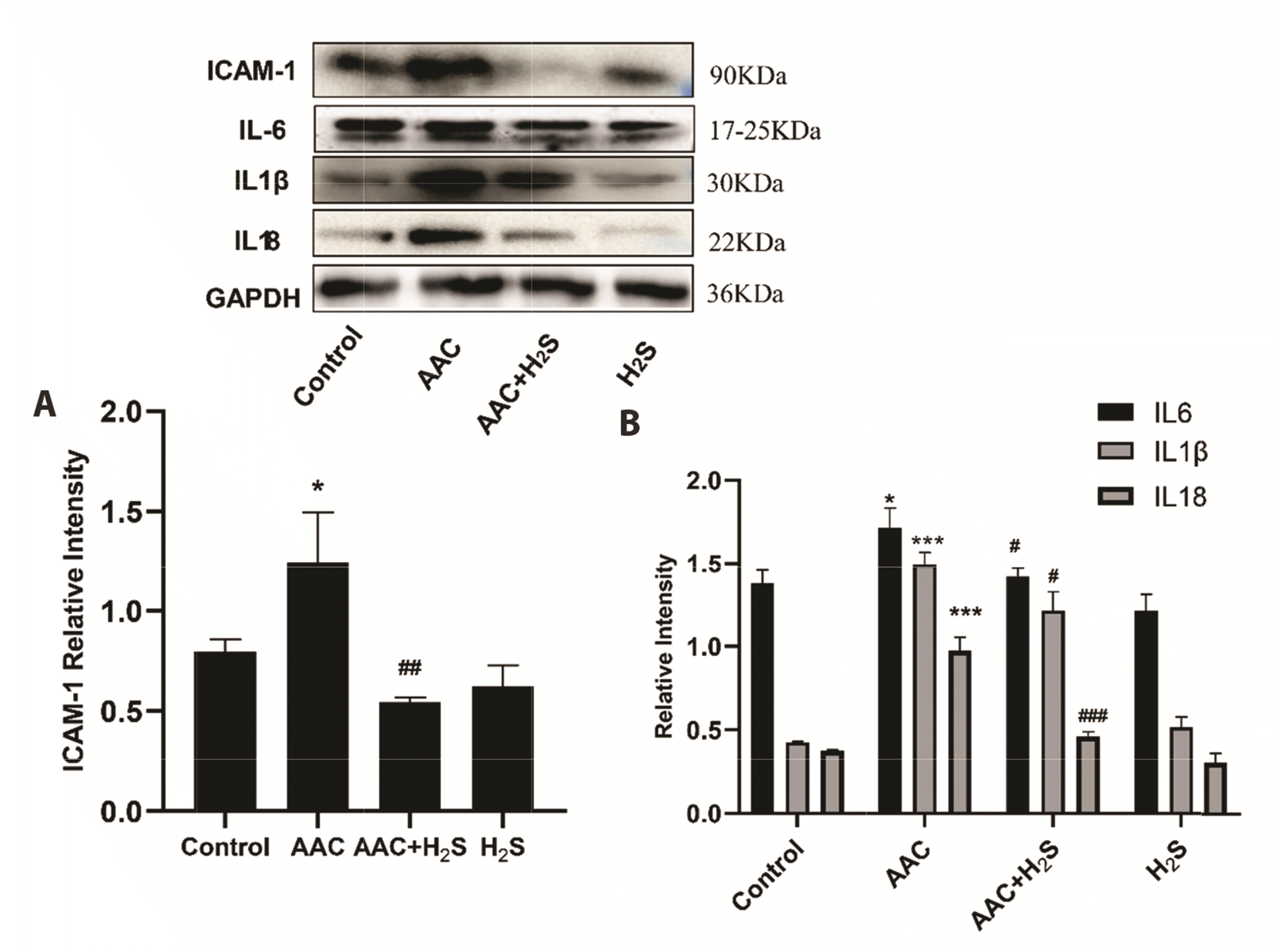 | Fig. 5Western blotting analysis the expression of (A) ICAM-1, (B) IL6, IL1β, IL18 in abdominal aorta coarctation (AAC)-induced myocardial fibrosis treated with control, AAC, AAC + H2S, and H2S.Values are presented as mean ± SD. H2S, hydrogen sulfide. *p < 0.05 vs. control, ***p < 0.001 vs. control, #p < 0.05 vs. AAC, ##p < 0.01 vs. AAC, ###p < 0.001.
|
The effect of H2S on the ultrastructure of myocardium in rats under the pressure overload
In this study, we observed the variations in ultrastructure of myocardium and autophagosomes in rats under the pressure overload. According to the results, myocardial fibers were closely arranged, and there were considerable mitochondria in the control group with no swelling or vacuolation. In comparison with the results of the control group, myocardial fibers were disorderly arranged, variations (e.g., mitochondrial swelling and vacuolation) were observed, and more autophagosomes could be observed in the field of vision in the AAC group. As compared with the results of the AAC group, myocardial fibers were arranged regularly in AAC + H2S group, mild edema occurred in myocardial fibers with no lytic necrosis, mitochondria were free from swelling or vacuolation, and less autopahgosomes were observed in the field of vision. In comparison with the results of the control group, the ultrastructure of myocardial tissue in H2S group were almost unchanged (Fig. 6).
 | Fig. 6Hydrogen sulfide (H2S) inhibitions abdominal aorta coarctation (AAC)-induced myocardial fibrosis mitophagy.Representative images of transmission electron microscopy (black arrows indicate mitophagy). Transmission electron micrographs at bars of 0.5 µm. (A) Control group. (B) AAC group. (C) AAC + H2S group. (D) H2S group.
|
The effect of H2S on the expression of autophagy-related proteins in myocardium of rats under the pressure overload
To observe the expression levels of autophagy-related proteins in myocardial tissue of rats under the pressure overload and the intervention effect of H2S. We used Western-blot method to detect the expression of P-eif2α, autophagy-related pathways, autophagy-related proteins. The results indicated that the protein expression levels of P-eif2α (p = 0.0398), ATG3 (p = 0.0282), ATG16L1 (p = 0.0112), Beclin1 (p = 0.0109), LC3A/B (p = 0.0430) in myocardium in AAC group increased significantly than the control group (Fig. 7), while those of P-PI3K (p = 0.0150), P-AKT1 (p = 0.0126) declined noticeably (Fig. 8). Compared with the results of the AAC group, the protein expression levels of P-eif2α (p = 0.0043), ATG3 (p = 0.0393), ATG16L1 (p = 0.0422), Beclin1 (p = 0.0357), LC3A/B (p = 0.0146) expression levels of these proteins in myocardial tissue in AAC + H2S group were significantly decreased, while those of P-PI3K (p = 0.0443), P-AKT1 (p = 0.0350) increased noticeably. In comparison with the results of the control group, the protein expression levels of P-eif2α (p = 0.6695), ATG3 (p = 0.0571), ATG16L1 (p = 0.3420), Beclin1 (p = 0.3308), LC3A/B (p = 0.2117), P-PI3K (p = 0.8407), P-AKT1 (p = 0.2335) in myocardial tissue of H2S group were almost unchanged, suggesting no differences with statistical significance.
 | Fig. 7Western blotting analysis the expression of (A) Atg3, (B) Atg16L1, (C) Beclin1, (D) LC3A/B in abdominal aorta coarctation (AAC)-induced myocardial fibrosis treated with control, AAC, AAC + H2S, and H2S.Values are presented as mean ± SD. H2S, hydrogen sulfide. *p < 0.05 vs. control, #p < 0.05 vs. AAC.
|
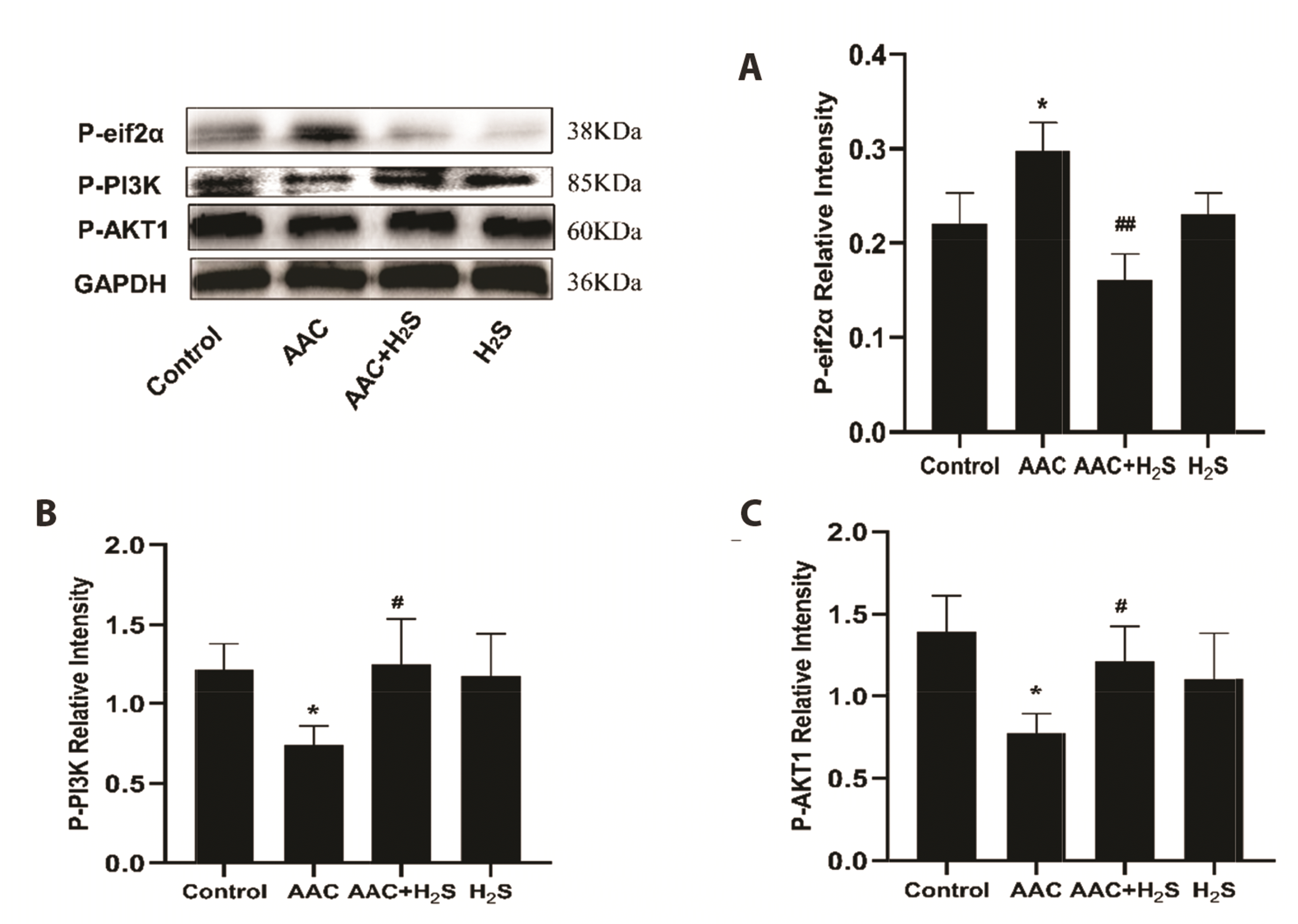 | Fig. 8Western blotting analysis the expression of (A) P-eif2α, (B) P-PI3K, (C) P-AKT1 in abdominal aorta coarctation (AAC)-induced myocardial fibrosis treated with control, AAC, AAC + H2S, and H2S.Values are presented as mean ± SD. H2S, hydrogen sulfide. *p < 0.05 vs. control, #p < 0.05 vs. AAC, ##p < 0.01 vs. AAC.
|
H2S inhibits Ang II-induced cardiomyocyte pyroptosis through down-regulation of eif2α phosphorylation
As we all know, γ-Cystathionase (CTH, EC: 4.4.1.1) is the key enzyme for the production of H2S so in our experiments we used Wertern blotting to detect the expression of CTH (Fig. 9). In order to further clarify the internal regulatory mechanism of H2S intervention and protection of pressure overload cardiomyopathy, this study used Ang II to induce an in vitro experimental model of H9c2 cardiomyocytes, and Western blotting was used to explore the effect of H2S on eif2α phosphorylated protein, PI3K/AKT1 signaling pathway and the expression of pyroptosis-related proteins. The results showed that compared with the control group, the expression of P-eif2α (p = 0.0280) in cardiomyocytes of the Ang II group was increased, the PI3K (p = 0.0483)/AKT1 (p = 0.0483) autophagy pathway was inhibited, and the pyroptosis-related proteins Caspase1 (p = 0.0469) and Caspase3 (p = 0.0212) were also significantly upregulated (Fig. 10). After H2S intervention, the expression of P-eif2α (p = 0.0084) was down-regulated, the PI3K (p = 0.0092)/AKT1 (p = 0.0092) autophagy pathway was activated, and the pyroptosis-related proteins Caspase1 (p = 0.0374) and Caspase3 (p = 0.0360) were also significantly down-regulated; the expression levels of the above proteins in the cardiomyocytes of the H2S and control groups did not change significantly, the difference was not statistically significant (p > 0.05). However, in the H2S intervention group, eif2α phosphorylation (p = 0.0029) activation (BTdCPU: 10 μm [18]) was added, and PI3K (p = 0.0092)/AKT1 (p = 0.0266) signaling pathway was obviously inhibited, and the pyroptosis proteins Caspase1 (p = 0.0316) and Caspase3 (p = 0.0466) were significantly up-regulated (Fig. 11).
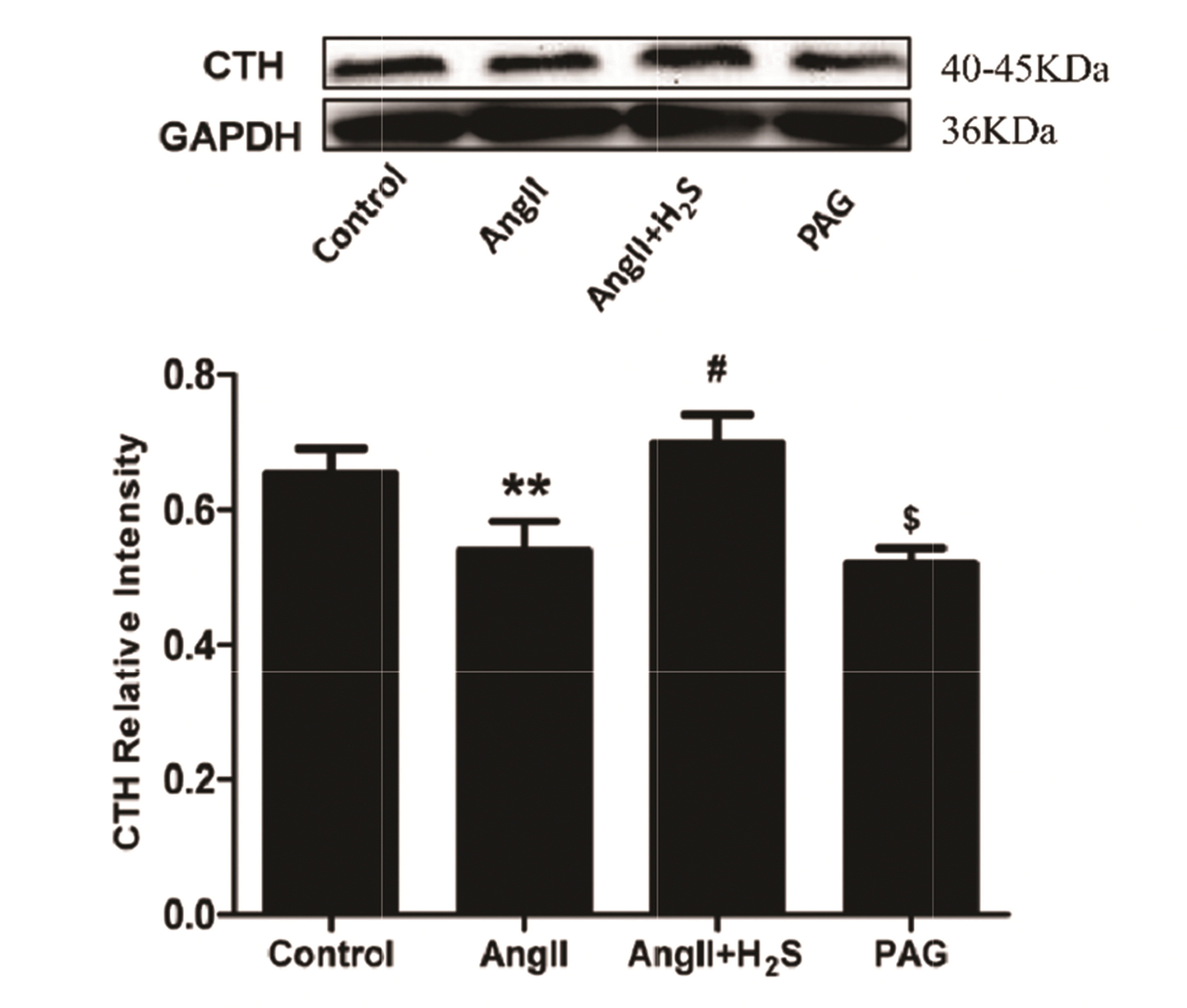 | Fig. 9Western blotting analysis the expression of CTH in H9c2 cells treated with control, Ang II, Ang II + H2S, and PAG.Values are presented as mean ± SD. CTH, γ-Cystathionase; Ang II, angiotensin II; H2S, hydrogen sulfide; PAG, DL-Propargylglycine. **p < 0.01 vs. control, #p < 0.05 vs. Ang II, $p < 0.05 vs. Ang II + H2S.
|
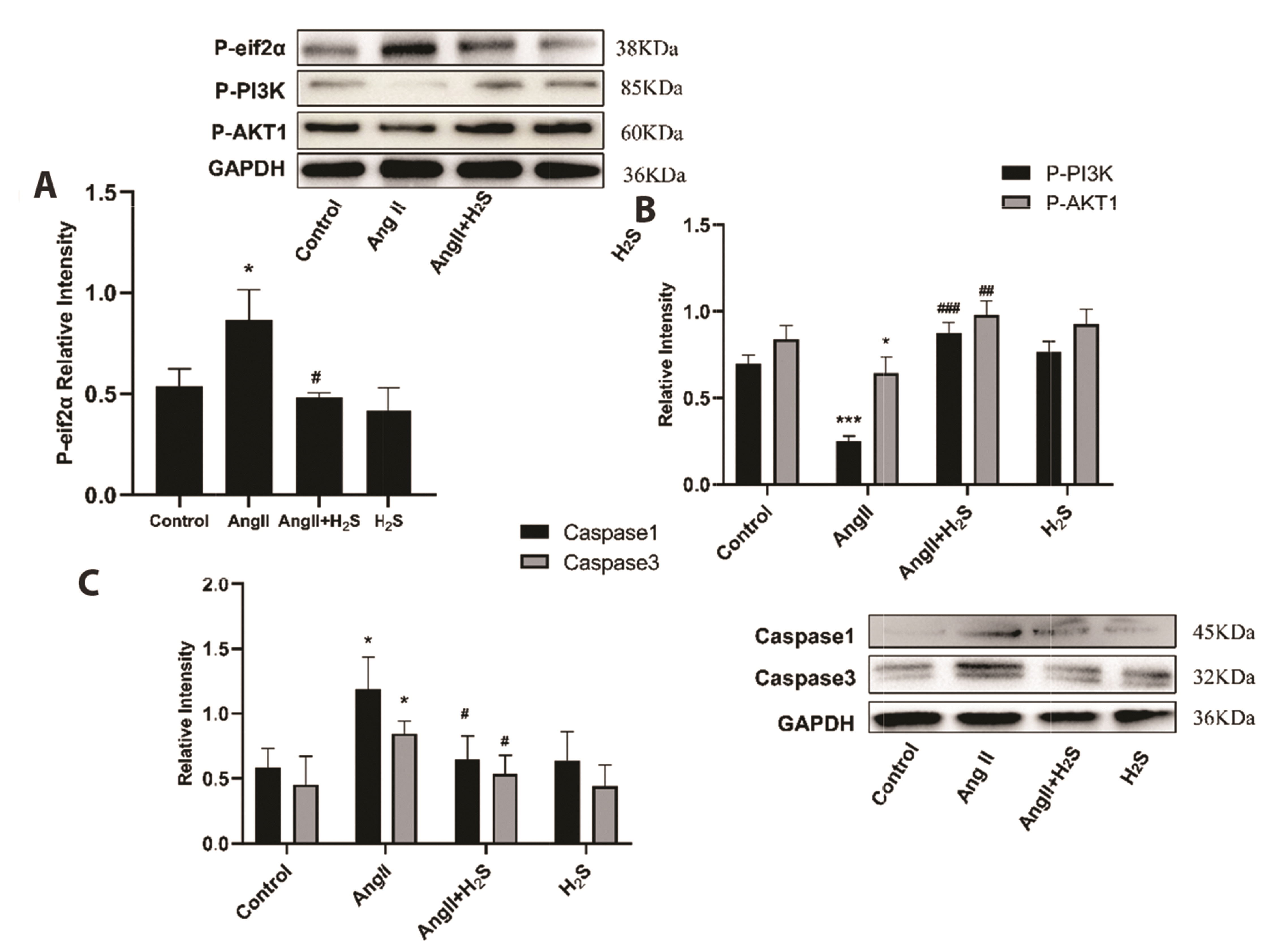 | Fig. 10Western blotting analysis the expression of (A) P-eif2α, (B) P-PI3K, P-AKT1, (C) Caspase1, Caspase3 in H9c2 cells treated with control, Ang II, Ang II + H2S, and H2S.Values are presented as mean ± SD. Ang II, angiotensin II; H2S, hydrogen sulfide. *p < 0.05 vs. control, ***p < 0.001 vs. control, #p < 0.05 vs. Ang II, ##p < 0.01 vs. Ang II, ###p < 0.001 vs. Ang II.
|
 | Fig. 11Western blotting analysis the expression of (A) P-eif2α, (B) P-PI3K, P-AKT1, (C) Caspase1, Caspase3 in H9c2 cells treated with control, Ang II, Ang II + H2S, Ang II + H2S + BTdCPU, and H2S.Values are presented as mean ± SD. Ang II, angiotensin II; H2S, hydrogen sulfide; BTdCPU, eif2α phosphorylation activation. *p < 0.05 vs. control, #p < 0.05 vs. Ang II, ##p < 0.01 vs. Ang II, $p < 0.05 vs. Ang II + H2S, $$p < 0.05 vs. Ang II + H2S.
|
Go to :
DISCUSSION
As people’s lifestyle has been changed, and aging of population is being accelerated, the incidence rate of hypertension shows an increase year by year. Also, hypertension is another major risk factor leading to cardiac failure and cardiovascular death. Statistics suggested that nearly half of cardiovascular deaths are clinically associated with hypertension, and cardiac failure is one of the underlying causes. The four-year mortality rate of cardiac failure reaches 50%, and pressure overload caused by hypertension may cause ventricular stiffness and cardiac dysfunction to enhance, which has been reported to show a close relation to MF in recent years [19]. Thus, reversing or postponing MF may be critical to enhance hypertension and prognosis of cardiac failure [20]. In this study, a model of rats under the pressure overload-induced MF was successfully built through AAC induced cardiomyocyte injury model. It was found that the hypertension of AAC group was obviously higher than that of the control group. By Masson staining, it was observed that deposition of collagen fibers obviously increased in myocardial interstitium of rats with AAC. Also, the results of Western blotting assay suggested that the expression of MMPs/TIMPs in myocardial tissue in rats under the pressure overload in AAC group was noticeably dysregulated. MMPs act as the major enzyme system that degrades extracellular matrix components, while TIMPs refer to endogenous specific inhibitors of MMPs. The interaction and dynamic balance between MMPs and TIMPs act as a critical factor to maintain ECM stability [21], and enhanced activity of MMPs disrupts the dynamic equilibrium of the ECM and causes collagen fibril deposition, therefore TIMPs, which are inhibitors of MMPs, increase negatively to achieve dynamic equilibrium [22], which is also consistent with our experimental results. Studies have also found that obvious MMPS/TIMPS dysregulation exists in rats under the pressure overload, and it is associated with excessive collagen deposition in myocardial interstitium and MF.
Pyrotosis is a kind of pro-inflammatory regulatory necrosis mode [23]. There are two main molecular mechanisms of pyroptosis: classical Caspase-1-dependent and non-Caspase-1-dependent pathways, both of which can lead to the release of pro-inflammatory cytokines IL-1β and IL-18 and expand local or systemic inflammation. Recent studies have also identified a new pathway of pyroptosis: activation of Caspase-3 by specific stimuli (originally Caspase-3 is a substrate for apoptosis), followed by Caspase-3-induced cleavage of Gasdermin E (GSDME) into C-terminal fragments (GSDME-CT) and N-terminal fragments (GSDME-NT), of which GSDME-NT is involved in pyroptosis [24,25]. Pyroptosis is a highly regulated cell death process, and in many cases, inhibition of this process by pharmacological or genetic intervention has a protective effect on the heart. Therefore, this process is a potential target for therapeutic intervention to prevent cardiovascular disease, and recent studies have also confirmed that cardiomyocyte pyroptosis is closely bound up with the occurrence of cardiovascular diseases, especially MF [5]. The Qiu team’s research [26] shows that inhibiting autophagy can reduce pyroptosisand ultimately improve non-alcoholic liver cirrhosis; this indicates that autophagy activation induces pyroptosis. Although, the study in Jiang [27] found that autophagy inhibits pyroptosis, which is contrary to previous reports. Based on the in-depth analysis of previous autophagy activation can induce myocardial remodeling after myocardial infarction, our laboratory believes that moderate autophagy refers to a process of self-salvation [6,7], excessive autophagy, cells will be damaged, so the apoptosis or death of cells will be promoted [8], this has been further explained in our study. We found that in AAC-induced SD rats, pyroptosis proteins Caspase1, Caspase3, inflammation-related proteins IL-6, IL-1β, IL-18, ICAM-1, the expression of autophagy-related proteins ATG3, ATG16L1, LC3A/B and Beclin1 was significantly increased, the PI3K/AKT1 signaling pathway was inhibited, and the formation of autophagosomes in SD rats induced by AAC was also observed by electron microscopy, indicating that AAC-induced autophagy in SD rats. After activation, the induced cell pyroptosis leads to pressure overload and MF, which may be related to the PI3K/AKT1 signaling pathway.
Eif2α mainly by binding to guanine triphosphate, provides promoter methionyl transfer RNA (Mett RNAi Met) to the small ribosomal subunit initiation complex, thereby participating in protein initiation phase synthesis [28]. Studies have pointed out that the level of phosphorylation of eif2α in response to various stresses is a key part of the overall stress response, which is called the integrated stress response [29,30]. Moreover, some studies have pointed out that eif2α phosphorylation activation blocks protein synthesis, and activated P-eif2α protein negatively regulates PI3K activity [14], which is considered a common factor in the occurrence of many diseases [13,31]. It is well known that PI3K/AKT1 signaling pathway is a classical autophagy pathway, and some studies have shown that renal denervation can improve myocardial remodeling induced by pressure overload, and its mechanism is related to the inhibition of autophagy. There are also studies showing that autophagy inhibitor 3-MA can improve left ventricular hypertrophy induced by pressure overload [32]. All these studies suggest that inhibiting excessive autophagy may be a key intervention to improve myocardial remodeling under pressure overload. In this study, it was also observed that the expression of P-eif2α was significantly increased and the PI3K/AKT autophagy pathway was inhibited in AAC group, which was also confirmed in H9c2 cardiomyocytes induced by Ang II. This study shows that pyrophagy is involved in the mechanism of pressure overload-induced MF and is associated with the inhibition of eif2-phosphorylated PI3K/AKT1 autophagy pathway.
H2S refers to the third gaseous signal molecule found after NO and CO, CSE is the key enzyme of endogenous H2S source in cardiovascular system [33], CBS and 3MST participate in the production of endogenous H2S in peripheral vascular tissue [34]. Several studies have reported that H2S have multiple biological effects on the cardiovascular system and have protecting effect on the heart. The previous research of the research group also showed that hydrogen sulfide can improve myocardial remodeling by reducing endoplasmic reticulum stress [35]. Some studies suggested that H2S can facilitate the process of left ventricular remodeling in rats with hypertension-induced heart failure [36], whereas the effect of H2S on AAC-induced MF in rats and the relevant regulatory mechanisms remain unclear. The results show that the heart function has not significantly decreased in the early stage of pressure overload, and we speculate that it may be in the compensatory phase at this time, however, significant changes in myocardial structure and abnormal molecular signal functions have been observed at this stage. After intervention with H2S donor, the deposition of collagen in myocardium in rats in AAC + H2S group under the pressure overload was obviously reduced, while MMPs/TIMPs dysregulation was noticeably improved, at the same time, it was found that it can significantly inhibit pyroptosis. In vitro experiments have also seen that H2S can inhibit Ang II-induced pyroptosis. Meanwhile, both in vitro and in vivo experiments showed that H2S could inhibit eif2α phosphorylation and activate the PI3K/AKT1 autophagy pathway. In order to further confirm whether hydrogen sulfide activates the PI3K/AKT1 autophagy pathway by inhibiting eif2α phosphorylation, in this study, after adding eif2α phosphorylation activation (BTdCPU: 10 μm [18] MCE, Shanghai, China) to the Ang II-induced cell injury model, it was clearly observed that pyroptosis inhibited in the H2S treatment group, and the PI3K/AKT1 autophagy pathway was inhibited, which suggests that hydrogen sulfide activates the PI3K/AKT1 autophagy pathway to improve cell pyroptosis by inhibiting eif2α phosphorylation.
In summary, this study found that H2S can activate the PI3K/AKT1 autophagy pathway by regulating eif2α phosphorylation expression, thereby inhibiting pyroptosis and improving pressure overload MF, however, more of these regulatory mechanisms remain to be further investigated. The results of this study are expected to provide new targets for intervention and treatment of hypertensive cardiomyopathy, as well as to develop novel exogenous H2S-releasing drugs to improve the occurrence and development of MF, and further open up the clinical application prospects of H2S-releasing drugs for anti-myocardial remodeling.
Go to :
 XML Download
XML Download