

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR: Hemostasis is a defense mechanism against vascular damage, caused by internal diseases or external injuries, that involves two different levels of damage (the endothelium alone or combined with extravascular tissue). In intravascular injury, generalized endotheliopathy may result from disease-induced endothelial damage, provoking systemic endothelial injury, which is observed in sepsis and other critical illnesses. Systemic endotheliopathy, sometimes presenting with thrombotic thrombocytopenic purpura (TTP)-like syndrome, causes “microthrombogenesis” and produces “microthrombi” strings by activating only the unusually large von Willebrand factor multimer (ULVWF) path, but not tissue factor path.
TTP-like syndrome produces pathological conditions of microvascular thrombosis similar to those found in TTP. However, TTP-like syndrome involves normal hemostasis with endothelial injury, whereas TTP involves pathological hemostasis without endothelial injury [1]. Herein, we report a case of TTP-like syndrome after COVID-19 (Coronavirus disease-19) vaccination.
CASE
A 73-year-old man with a history of laparoscopic-assisted proximal gastrectomy for early gastric cancer presented with generalized edema, thrombocytopenia, and azotemia. After the first dose of the AstraZeneca (AZ) vaccine, he underwent routine check-ups, and grade 3 proteinuria and microscopic hematuria were detected. At that time, the patient had a normal platelet count. Three days after the second dose of AZ vaccine, which was administered 12 weeks after the first dose, the patient developed acute gastroenteritis and received conservative therapy. Three weeks after the injection, the patient was transferred to the emergency room because of generalized edema and thrombocytopenia.
Initially, the patient’s blood test showed bicytopenia: WBC, 5,810/μL (normal range, 4–10×103/μL); Hb, 8.7 g/dL (normal range, 15–17 g/dL); and platelet, 16,000/μL (normal range, 150–400×103/μL) with MCV, 96.7 fL; MCH, 31.6 pg; MCHC, 32.7 g/dL; and reticulocyte, 4.41%. Chemistry findings were as follows: BUN, 72 mg/dL; creatinine, 1.32 mg/dL; total protein, 4.7 g/dL; albumin, 2.7 g/dL; total bilirubin, 2.04 mg/dL; direct bilirubin, 0.38 mg/dL; AST/ALT, 76/40 IU/L; ALP/γGT, 63/15 IU/L; LDH, 575 IU/L (normal range, 100–225 IU/L). Coagulation panel tests were normal except for increased D-dimer levels: PT INR, 0.99; aPTT, 32 s; fibrinogen, 248 mg/dL; D-dimer, 1.77 μg/mL. A peripheral blood smear showed schistocytosis of grade 2 (2–3 per high-power field of view) and severe thrombocytopenia. There was no evidence of thrombosis or active bleeding on brain and abdominal computed tomographic angiography. The PLASMIC score was 6 points (platelet count <30,000/μL, hemolysis; neither active cancer nor history of transplant; INR <1.5; creatinine <2.0 mg/dL) at that time.
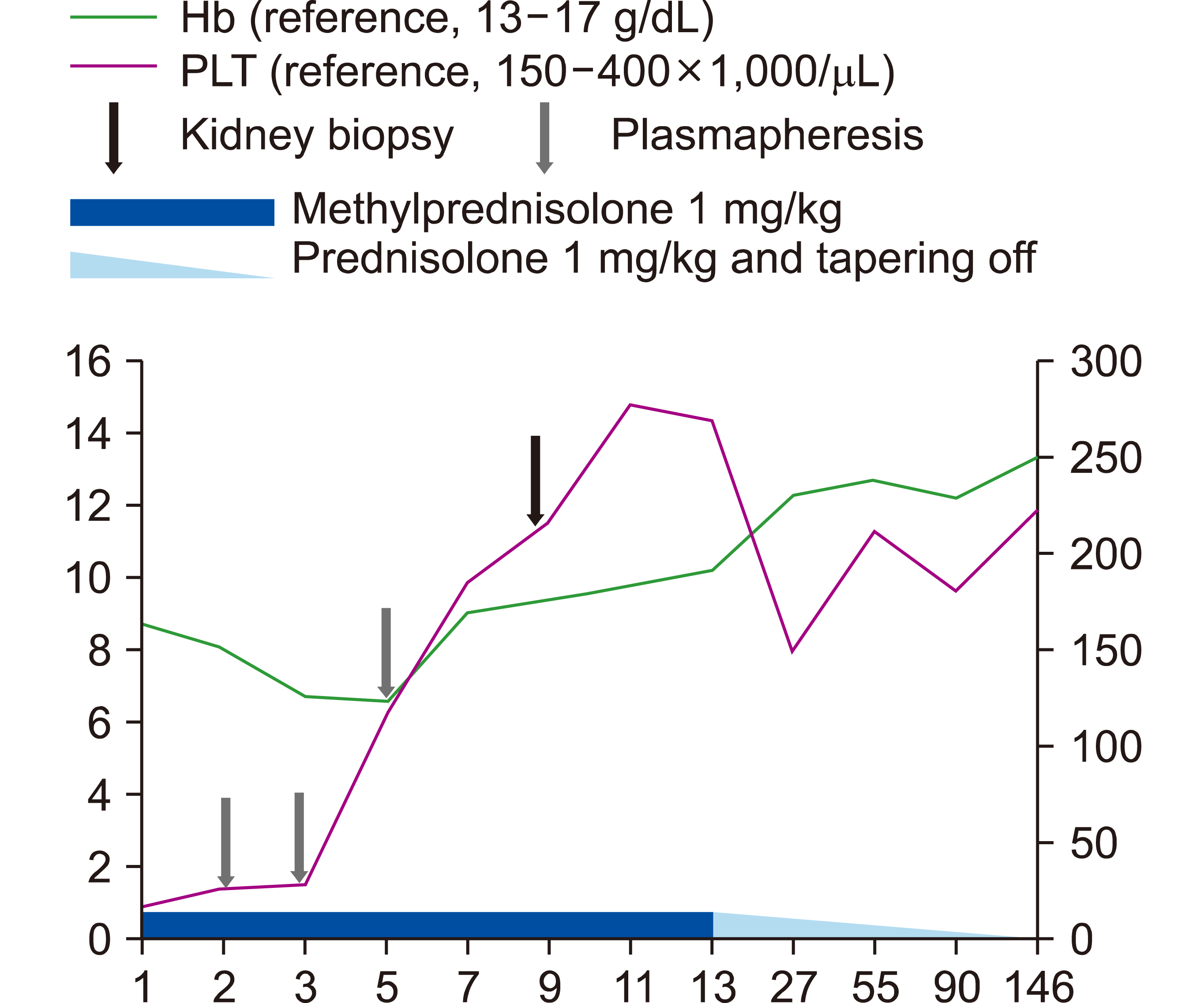
Plasmapheresis was initiated owing to a high suspicion of TTP, and methylprednisolone (1 mg/kg) was administered. After the third round of plasmapheresis, platelet counts and creatinine levels were normalized, and kidney biopsy was performed while continuing high-dose steroids (Fig. 1).
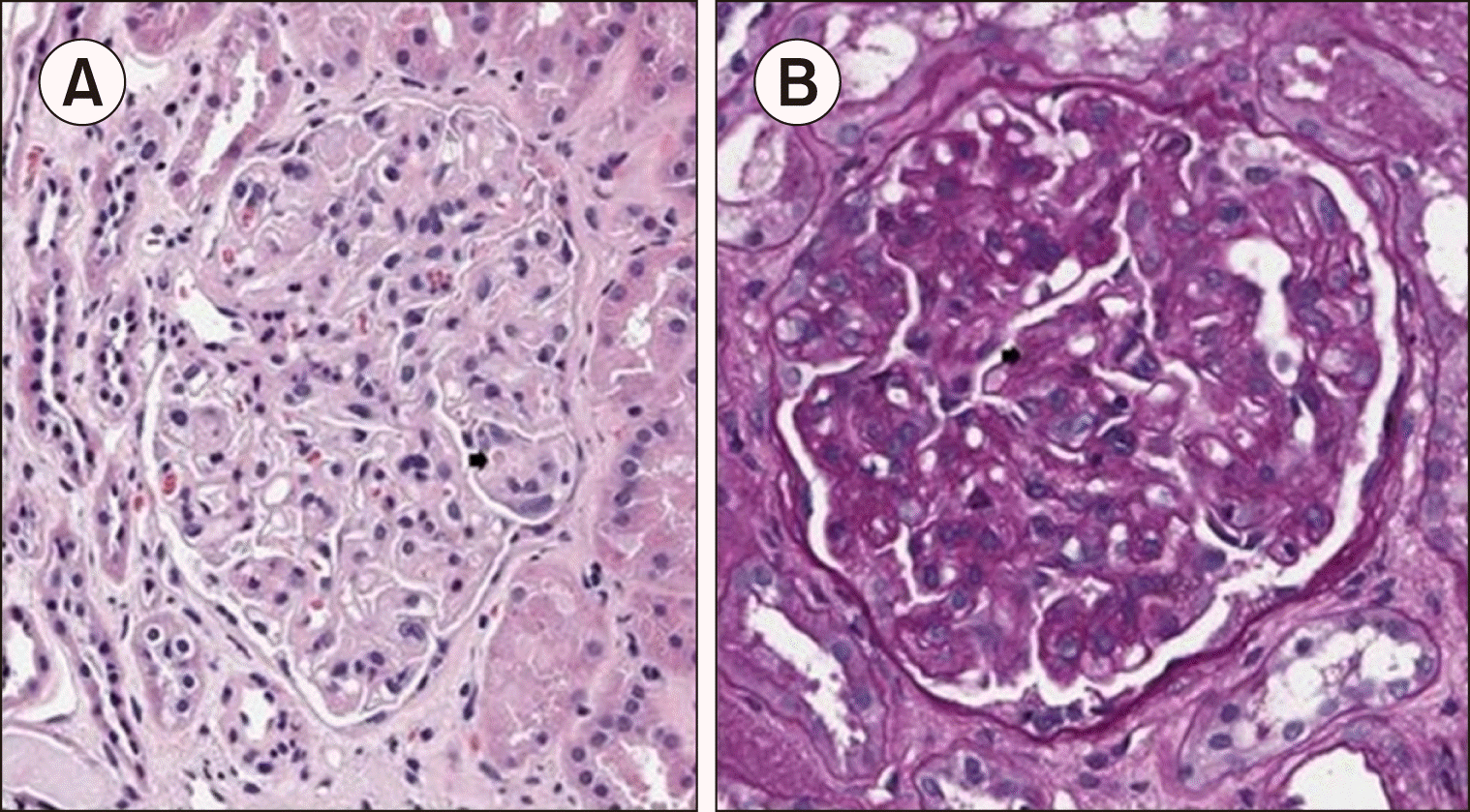
The initial ADAMTS 13 activity before plasma exchange was confirmed to be in the normal value of 46.8% (normal range, >40%), and the stool test for E. coli O157 was also negative. Kidney biopsy revealed endothelial swelling and some intraglomerular thrombi (Fig. 2A), segmental occlusion of the glomerular capillary lumen, and thickening of the shrunken capillary wall with double contours (Fig. 2B), consistent with thrombotic microangiopathy. Genetic mutations associated with atypical hemolytic uremic syndrome (HUS) were not identified using next-generation sequencing.
The steroid dose was tapered over five months, after which steroid administration was discontinued. Subsequently, complete remission with normal hemoglobin levels, platelet counts, and kidney function was maintained for 16 months.
DISCUSSION
TTP and TTP-like syndrome share similar pathological findings and microvascular hemolytic anemia. TTP is associated with a deficiency of ADAMTS 13 or the presence of autoantibodies to ADAMTS 13; in contrast, TTP-like syndrome may be observed in patients with severe diseases such as sepsis, trauma, and cancer [2]. Damage to the vascular endothelium increases the release of ULVWF and produces "microthrombi" that comprise a complex of platelets and ULVWF [3-5]. The clinical presentation often includes multiple organ dysfunction. In this case, HUS was ruled out based on a negative E. coli O157 stool test [6], and TTP could be ruled out due to the initial normal activity of ADAMTS 13. Because the patient was an older adult and had no aHUS-related genetic variation, aHUS was also a diagnosis that could be excluded. There were no episodes of major infection, trauma, or recurrent tumor, and the COVID-19 vaccine was believed to be a major recurrent stimulus that caused endothelial damage, leading to a TTP-like syndrome. At that time, vaccine-induced immune thrombocytopenia and thrombosis syndrome (VITT) after adenoviral vector vaccination was a major issue; therefore, this diagnosis needed to be excluded. Patients with VITT should have objectively proven arterial and/or vein thrombosis accompanied by thrombocytopenia caused by the heparin PF4 autoantibody. In a prospective cohort study in the United Kingdom on the clinical characteristics of VITT, the overall mortality rate was 22% among 294 patients with definite or probable VITT, and the risk factors for death in these patients were lower platelet counts, intracranial hemorrhage, and cerebral venous sinus thrombosis [7].
However, this patient did not develop overt thrombosis during the clinical course and showed rapid improvement after short-course plasmapheresis and immunosuppression without additional anticoagulants. Therefore, he was diagnosed with TTP-like syndrome after adenoviral vector vaccination.
In conclusion, TTP-like syndrome should be considered for patients who present with thrombocytopenia with microangiopathic hemolytic anemia after any unknown or rarely reported stimuli, including COVID-19 vaccination. We believe that this case report will deepen our understanding of vaccine-related immunological and vascular responses. In our patient, plasma exchange and high-dose corticosteroids reversed the critical condition of TTP-like syndrome after adenoviral vector vaccination. However, the exact pathogenesis of TTP-like syndrome after COVID-19 adenoviral vector vaccination has not yet been established. Therefore, further research is needed to reveal its pathogenesis.
 XML Download
XML Download