

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION

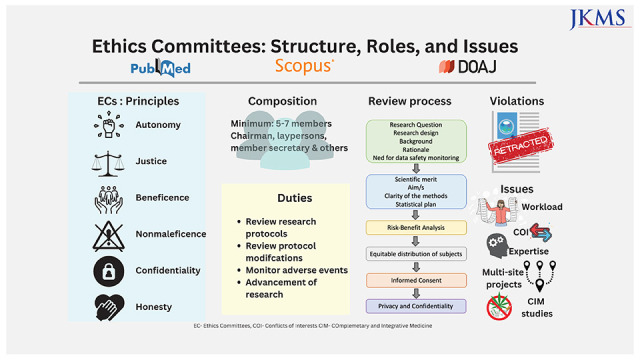
1. Autonomy: respect the patient’s right to act on his/her own value and choice.
2. Justice: fair treatment of the research subjects.
3. Beneficence: work for the benefit of the patient.
4. Nonmaleficence: primum non-nocere or first do no harm to the patient.
5. Confidentiality: privacy protection.
6. Honesty: truthfulness in terms of the study.
METHODS
RESEARCH, SUBMISSION PROCESS, AND EXEMPTIONS
Table 1
Type of research studies and approvals/exemptions

EC PERSONNEL, THEIR EXPERIENCE, AND DUTIES
Personnel
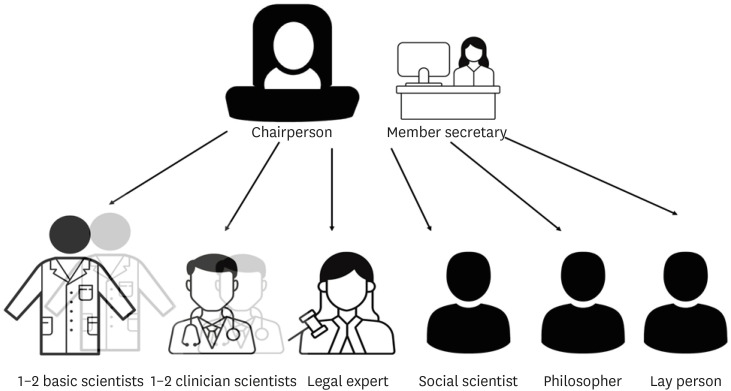
a) Most countries in Europe, the USA, and South Korea have a requirement of at least five members whereas recommendations in China and India need a minimum of seven members and a maximum of 12–15 members.1436373839
b) At least one member who is autonomous, independent of the institution or trial site. It is mandatory that the chairperson of the EC is not part of the institution where the research is to be conducted.
c) At least one member who is from the non-scientific community (Fig. 2).
Responsibilities
Duties
• The IRB should obtain and review all the necessary documents for the research/trial within a reasonable time and document its views following standardized operating procedures with clear identification of the dates for approval, modifications, disapproval, or termination of an ongoing trial that was initially approved in writing.
• Qualification of the investigators should be considered for the proposed research.
• Reviewing of ongoing research as appropriate to the risks involved (at least once a year).
• Protocols indicating exemption of prior consent of the subject or their legally acceptable representative (e.g., emergency situations) should be assessed in detail for all the regulatory needs.
• Review the sum and method of compensatory payment to subjects if required.
• Functions should be performed as per written SOPs which should comply with the GCP guideline.
Table 2
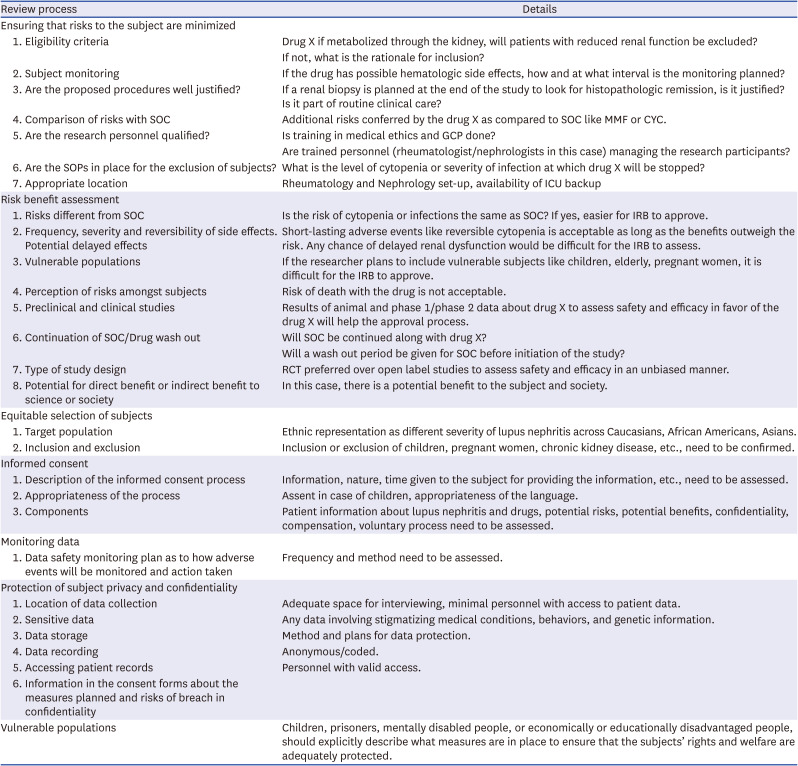
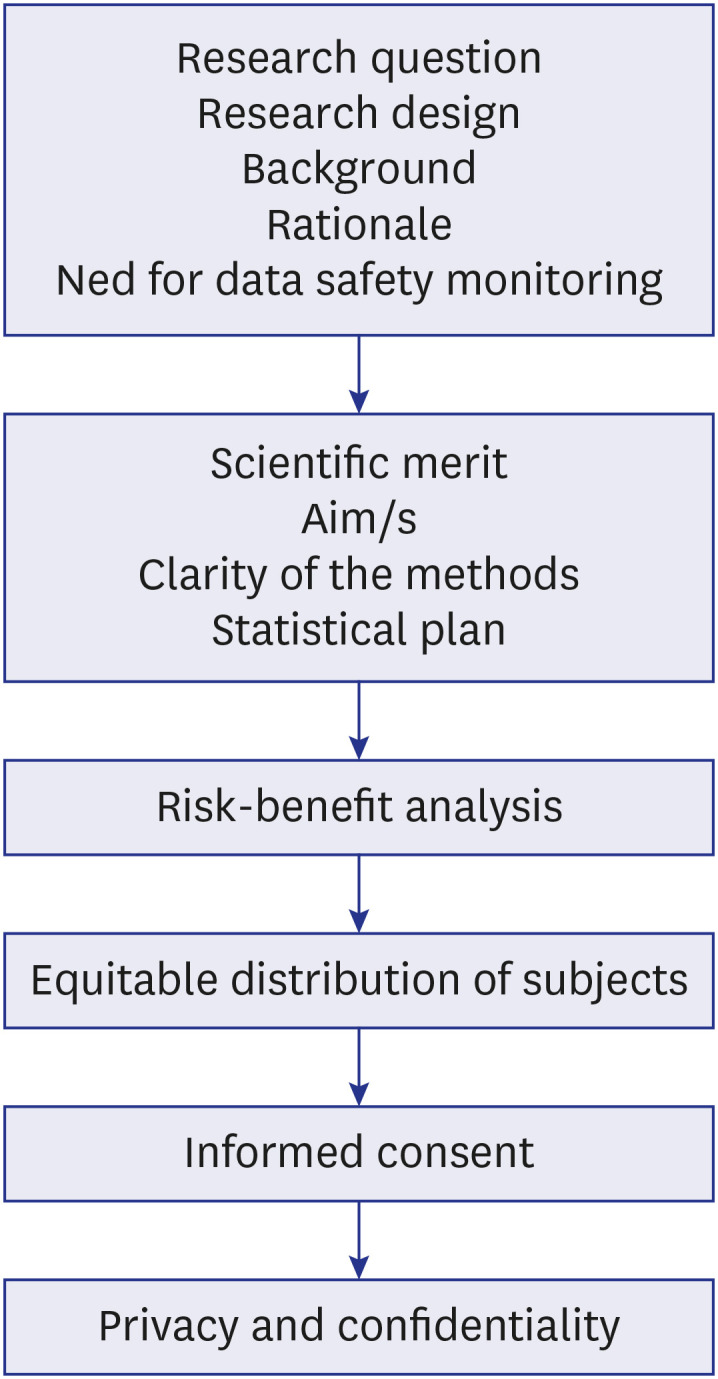
Review process of a study by the Ethics Committee8
RISKS, BENEFITS, CONFIDENTIALITY, AND PRIVACY ISSUES IN RESEARCH PROTOCOLS
• Review the consent document and assess the sensitivity of the information, the duration for which it will be held, the usefulness of the information, and the ability to protect it.
• For multicenter projects, review the measures taken by the research team to maintain the privacy of the research subjects including the number of personnel with access to the information, data storage, and transfer.
• An ongoing review of the research must include monitoring of confidentiality issues to check for maintenance of the same and the need for a revised privacy protection plan.
• Educate researchers and IRB members regarding the data privacy and protection process.46
VIOLATIONS OF ETHICS APPROVAL RULES AND REGULATIONS
Table 3
Subject wise analysis of examples of retractions secondary to lack of IRB approvals/ethical issues
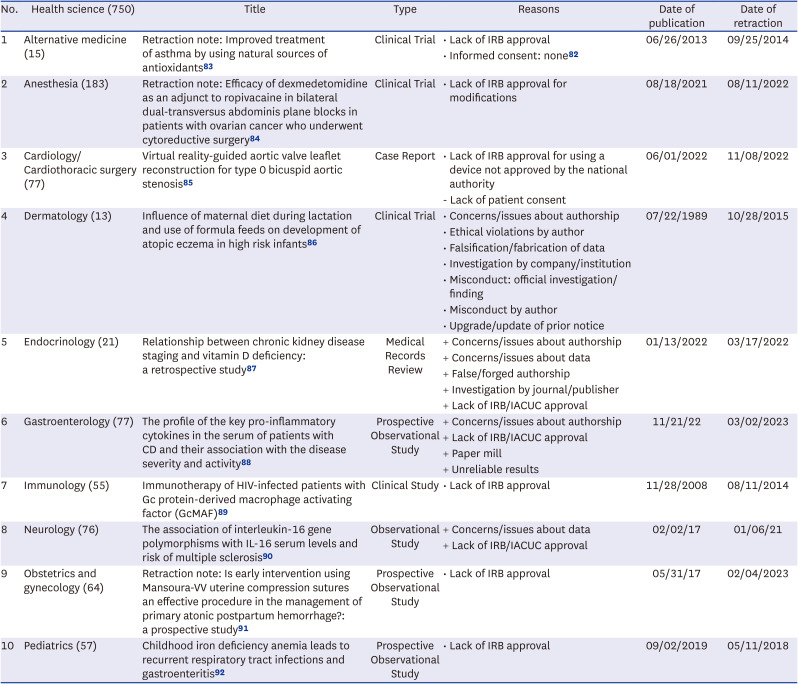
| No. | Health science (750) | Title | Type | Reasons | Date of publication | Date of retraction |
|---|---|---|---|---|---|---|
| 1 | Alternative medicine (15) | Retraction note: Improved treatment of asthma by using natural sources of antioxidants83 | Clinical Trial | • Lack of IRB approval | 06/26/2013 | 09/25/2014 |
| • Informed consent: none82 | ||||||
| 2 | Anesthesia (183) | Retraction note: Efficacy of dexmedetomidine as an adjunct to ropivacaine in bilateral dual-transversus abdominis plane blocks in patients with ovarian cancer who underwent cytoreductive surgery84 | Clinical Trial | • Lack of IRB approval for modifications | 08/18/2021 | 08/11/2022 |
| 3 | Cardiology/Cardiothoracic surgery (77) | Virtual reality-guided aortic valve leaflet reconstruction for type 0 bicuspid aortic stenosis85 | Case Report | • Lack of IRB approval for using a device not approved by the national authority | 06/01/2022 | 11/08/2022 |
| - Lack of patient consent | ||||||
| 4 | Dermatology (13) | Influence of maternal diet during lactation and use of formula feeds on development of atopic eczema in high risk infants86 | Clinical Trial | • Concerns/issues about authorship | 07/22/1989 | 10/28/2015 |
| • Ethical violations by author | ||||||
| • Falsification/fabrication of data | ||||||
| • Investigation by company/institution | ||||||
| • Misconduct: official investigation/finding | ||||||
| • Misconduct by author | ||||||
| • Upgrade/update of prior notice | ||||||
| 5 | Endocrinology (21) | Relationship between chronic kidney disease staging and vitamin D deficiency: a retrospective study87 | Medical Records Review | + Concerns/issues about authorship | 01/13/2022 | 03/17/2022 |
| + Concerns/issues about data | ||||||
| + False/forged authorship | ||||||
| + Investigation by journal/publisher | ||||||
| + Lack of IRB/IACUC approval | ||||||
| 6 | Gastroenterology (77) | The profile of the key pro-inflammatory cytokines in the serum of patients with CD and their association with the disease severity and activity88 | Prospective Observational Study | + Concerns/issues about authorship | 11/21/22 | 03/02/2023 |
| + Lack of IRB/IACUC approval | ||||||
| + Paper mill | ||||||
| + Unreliable results | ||||||
| 7 | Immunology (55) | Immunotherapy of HIV-infected patients with Gc protein-derived macrophage activating factor (GcMAF)89 | Clinical Study | • Lack of IRB approval | 11/28/2008 | 08/11/2014 |
| 8 | Neurology (76) | The association of interleukin-16 gene polymorphisms with IL-16 serum levels and risk of multiple sclerosis90 | Observational Study | + Concerns/issues about data | 02/02/17 | 01/06/21 |
| + Lack of IRB/IACUC approval | ||||||
| 9 | Obstetrics and gynecology (64) | Retraction note: Is early intervention using Mansoura-VV uterine compression sutures an effective procedure in the management of primary atonic postpartum hemorrhage?: a prospective study91 | Prospective Observational Study | • Lack of IRB approval | 05/31/17 | 02/04/2023 |
| 10 | Pediatrics (57) | Childhood iron deficiency anemia leads to recurrent respiratory tract infections and gastroenteritis92 | Prospective Observational Study | • Lack of IRB approval | 09/02/2019 | 05/11/2018 |
ISSUES AND ONGOING DEVELOPMENTS
• Composition: Most studies indicate a skewed gender representation in the structure IRBs. Further, the participation of laypersons on the board is minimal.1442555657
• Overburdened IRBs, delays, and operational costs: The IRB reviews have been associated with delays from over 4 to 7 months on average from surveys conducted across the USA.5859 A delay in biomedical research can translate into more than monetary loss as biomedical research saves lives and a delay in the approvals can result in greater loss of life.60 An older survey conducted across 63 institutions (with 20 being low volume, 24 intermediate volume, and 19 being high volume centers) in the USA in 2005 reported the median amount spent by academic medical centers on IRB was $750,000/year with an average of $559 per review. The main costs are divided across staff salary, board salary, space, outsourcing of the reviews, travel, supplies, and equipment.61 Over the years, there is a definite increase in the number of ongoing research projects thus increasing these costs further. Furthermore, documentation of Food and Drug Administration (FDA) warning letters to IRBs was predominantly related to paperwork stressing on documentation of reviews and meetings rather than ethical issues.62 Increasing paperwork further results in delays and added costs. These deficiencies are more marked in developing nations like India and China dealing with issues like lack of regulation, informal ethics reviews, lack of supervision, and insufficient ethics review capacity.6364
• Multi-site projects: With multicenter projects on the rise, a single protocol is often reviewed by multiple IRBs. In a review of 17 articles reported from UK, USA and Europe, which underwent multiple IRB reviews of the same protocol there were discrepancies in the judgment. Five of 26 reported rejection at some and acceptance by some IRBs. However, there were great differences in the protocol revisions, consent, patient information sheets, risk-benefit assessment, and compensation arrangements.65 Keeping these issues in mind, the Common Rule in the USA was revised in 2017 with IRB approval required only from one center for multisite projects.66 This may be extrapolated to other nations or consideration of an expedited review at other sites when fully reviewed at one IRB can be considered.
• Independent EC and IEC: Independent ECs have inherent tissues of limitation of knowledge about the local community and use of these may promote IRB shopping. Whereas, local IRBs can have conflicts of interest as colleagues of investigators may be on the review board. Thus, a central IRB can alleviate some of these concerns by avoiding repetitive reviews, minimizing conflicts, and establishing a centralized adverse event reporting system.676869 A central IRB can be formed by experts on a particular subject or by a group of institutes like the National Cancer Institute’s Central IRB and the Biomedical Research Alliance of New York respectively.7071
• Scientific expertise of the IRB reviewers: The IRB reviewers may lack the scientific expertise to review sophisticated research projects that may affect the quality of the research.11145772 Regular training in research ethics and GCP along with adequate consultations with external experts is needed. This can be done at a national, regional, and international level. First, by identifying core issues and then solutions for them by focused training.73 Training of EC members is conducted across Central Asia and Eastern Europe under the framework of Forum for Ethics Committees in the Confederation of Independent States and Strategic Initiative for Developing Capacity in Ethical Review program that train members regarding GCP, bioethics, the establishment of an EC, review processes and SOPs, choosing independent consultants, and confidentiality agreements.74
• Review of studies involving complementary and integrative medicine (CIM) is a challenge due to the lack of quality evidence to support the basis for their use. Moreover, most international regulatory bodies and research regulations do not address CIM, thus leaving the review process and decision-making to the IRBs. However, it is to be emphasized here studies irrespective of the type (modern or CIM) must be reviewed using the same principles of respect, beneficence, and justice. Well-designed studies on CIM are essential to ascertain the health and safety of patients.75
DSMB
• The study outcome is such that a highly encouraging or detrimental result is a possibility in an interim analysis that may require an early termination of the study on ethical grounds.
• When the safety concerns are high, e.g., invasive therapy is administered.
• Previous data suggesting serious toxicity with the study treatment.
• Studies involving vulnerable populations.
• Studies including subjects at an increased risk of death or serious outcomes.
• Large, multisite, long-duration studies.
• To uphold participant safety.
• Ensure credibility and integrity of the trial for future subjects.
• Ensure the timely conclusion of the study so that the results can be disseminated.
• Identify protocol violations if any.
• Identify unexpectedly high dropouts and evaluate for the same.
• Ensure the validity of the results.
 XML Download
XML Download