

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Megalencephaly-capillary malformation-polymicrogyria syndrome (MCAP) is a rare genetic disorder that manifests as progressive megalencephaly/hemimegalencephaly with polymicrogyria, body overgrowth (segmental overgrowth or hemihypertrophy), cutaneous capillary malformation, and distal limb anomalies such as syndactyly or polydactyly [1]. Before the early 2010s, clinical features satisfying diagnostic criteria were the only way to diagnose this disease [1-3]. However, Mirzaa et al. [4] suggested that de novo germline mutation or postzygotic mutations in the phosphoinositide 3-kinase (PI3K)-AKT pathway played a crucial role in megalencephaly syndromes. Subsequently, it was reported recently that MCAP is a phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA)-related disorder characterized by overgrowth and vascular malformation caused by the PIK3CA gene mutation [5,6]. The disease course for MCAP is progressive in most cases, so early detection is crucial. In addition, tumor development is possible, even though its incidence is rare [5,7]. Moreover, targeted therapies, such as the PI3K-specific inhibitor alpelisib (Norvatis Pharmaceuticals Corporation), which was developed for breast cancer, are now being tested in MCAP cases and have shown initial successful outcomes in patients with PIK3CA-related disorders [8]. Such therapies are only applicable to a confirmed genetic diagnosis. Therefore, early genetic diagnosis of MCAP is becoming increasingly important. However, to our knowledge, there have been no reports in Korea of a patient receiving a genetic diagnosis of a PIK3CA gene mutation during the neonatal period. We report a patient who satisfied the clinical diagnostic criteria for MCAP and was confirmed to have a PIK3CA gene mutation during the neonatal period.
Go to : 
CASE REPORT
A female neonate born at 39 weeks and four days of gestation with a birth weight of 3,940 g via vaginal delivery in a local obstetrics clinic was referred to the neonatal intensive care unit (NICU) for post-resuscitation care. Prenatal ultrasound revealed no specific findings, except for macrocephaly. During birth, labor dystocia occurs because of macrocephaly. The infant was hypotonic and could not breathe spontaneously without crying. The patient’s respiration and activity improved after 15 minutes of positive pressure ventilation with a mask, and she was transferred to our NICU without any respiratory support.
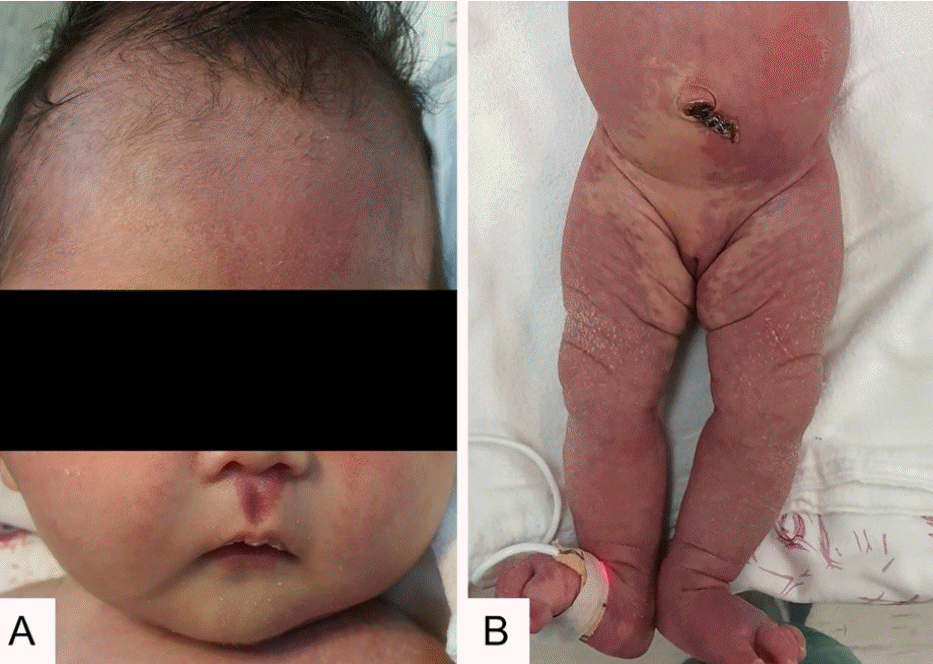
Physical examination revealed macrocephaly with a 51.5 cm head circumference (>97th percentile). Strawberry-colored patches were observed on the philtrum and lower lip, and widespread reticulated port-wine staining was observed on the forehead, trunk, and extremities. She had slightly smaller 2nd fingernails on both hands, and body asymmetry and somatic overgrowth were not observed (Figure 1). The initial arterial blood gas analysis showed mild metabolic acidosis (pH 7.3, pCO2 29 mm Hg, pO2 67 mm Hg, bicarbonate 15.6 mmol/L, base deficit 8.8), and the serum aspartate aminotransferase/alanine aminotransferase was 254/39 IU/L.
On the 2nd day after birth, clonic seizures with oxygen desaturation were observed for one minute. Serum lactate dehydrogenase and creatine kinase increased to 1,617 and 12,989 U/L, respectively. Brain magnetic resonance imaging (MRI) revealed left hemimegalencephaly with polymicrogyria in both the perisylvian and frontal gyri. The cavum septum pellucidum and vergae were also observed (Figure 2A). Electroencephalography (EEG) showed a normal sleep pattern. Abdominal ultrasonography revealed no abnormalities. Sanger sequencing was performed on the buccal mucosa sample after 20 days of age, and MCAP was confirmed with the PIK3CA mutation [c.1635G>T (p. Glu545Asp)]. The PIK3CA mutation was not identified in peripheral blood.
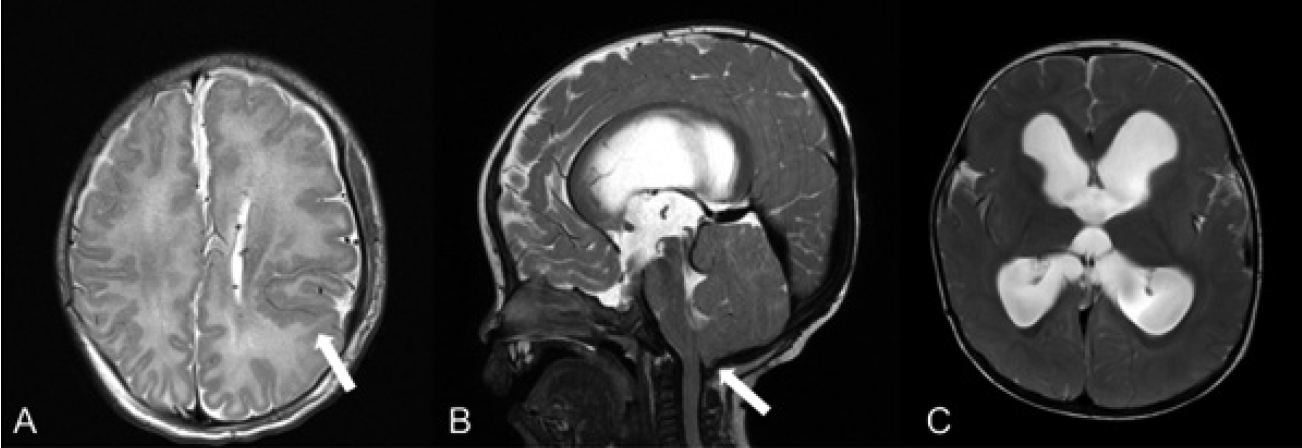
A follow-up brain MRI at 7 months of age revealed bilateral hydrocephalus secondary to progressive cerebral/cerebellar enlargement, which was associated with increased intracranial pressure (ICP). Cerebellar tonsillar herniation and thick corpus callosum were observed. (Figure 2B, C). Papilledema was also detected on retinal examination. A follow-up EEG revealed focal and generalized seizures. There were very frequent, nearly continuous, sharp, or spiked discharges from the central areas during sleep and intermittent polyspike discharges from both parieto-occipital areas. Additionally, several episodes of diffuse or bilateral slow-wave bursts were observed on the EEG. A ventriculoperitoneal (VP) shunt was surgically placed to alleviate increased ICP caused by hydrocephalus and cerebellar tissue overgrowth. For possible tumor conditions, abdominal ultrasonography and tumor markers, including human chorionic gonadotropin and alpha-fetoprotein, were regularly checked, and the results were all negative. She presented with moderate global developmental delay diagnosed using the Bayley Scales of Infant Development-II at 3 years of age (mental developmental index=62 and psychomotor developmental index <50). She was treated with rehabilitation therapy for language and motor developmental delay and laser therapy for skin lesions. The patient is currently on a waitlist for alpelisib treatment.
Go to :
DISCUSSION
To our knowledge, this is the first report in Korea of a patient receiving a genetic diagnosis of the PIK3CA gene mutation during the neonatal period. In Korea, Choi et al. [9] and Park et al. [10] reported patients clinically diagnosed with MCAP without genetic evaluation. Kim et al. [11] reported patients with lateralized overgrowth, including infants with MCAP and PIK3CA mutations. Park et al. [12] reported 12 patients diagnosed with MCAP and PIK3CA gene mutations during childhood.
MCAP is an overgrowth spectrum featuring megalencephaly and capillary malformation [1]. Antenatally, macrocephaly is the only clinical feature of MCAP, but after birth, cutaneous capillary malformation and distal limb anomalies can help in the diagnosis. Megalencephaly is accompanied by hydrocephalus, polymicrogyria, ectopic cerebellar tonsils, and thick corpus callosum. Capillary skin malformations are either midfacially localized in the philtrum and lips or are reticular and port-winecolored in the extremities and trunk. Affected patients also have distal limb anomalies with syndactyly, segmental overgrowth or hemihypertrophy, joint hypermobility, and thick doughy subcutaneous tissue [1-3]. Since MCAP was initially described in 1997 [13], several diagnostic criteria have been introduced [2,3]. Mirzaa et al. [1] established new diagnostic criteria in 2012, emphasizing the importance of perisylvian polymicrogyria. Our patient satisfied the diagnostic criteria; in particular, the brain anomaly was so significant that all criteria features were detected [1-3]. Brain anomalies can cause seizures, developmental delays, and hypotonia [14].
In most cases, MCAP progresses. Aggravating hydrocephalus can occur secondary to progressive cerebral or cerebellar overgrowth [1,15]. In our patient, the hydrocephalus was markedly aggravated; therefore, an emergent VP shunt was performed. Therefore, early diagnosis is important, and regular follow-up is essential because MCAP has extremely dynamic clinical symptoms.
Second, early genetic diagnosis is important because targeted gene therapy is becoming possible, and genetic therapy requires a genetic diagnosis. The PIK3CA gene mutation activates the PI3K/AKT/mammalian target of rapamycin (mTOR) pathway, a critical cell signaling pathway that regulates various cellular functions, including growth, proliferation, and metabolism [6]. Overactivation of this pathway is involved in cancer development or overgrowth syndrome [16]. The somatic mosaic mutation of the PIK3CA gene is associated with MCAP [4,12]. Targeted therapy to the PI3K/AKT/mTOR signaling pathway, which was originally developed as an anticancer therapy, has shown favorable outcomes in the reduction of tissue overgrowth, vascular lesions, and other functional complications in PIK3CA-related disorders [8]. Seizure, cognitive function, and behavior can be improved in MCAP by targeted therapy [8,17]. Alpelisib has not yet been approved by the Ministry of Food and Drug Safety (MFDS) for PIK3CA-related disorders in Korea. Kim et al. [11] reported two patients (one child and one adult) with PIK3CA-related disorders with the PIK3CA gene mutation who were treated with alpelisib for 18 months and showed alleviation of overgrowth of the lower legs without adverse effects, including hyperglycemia, rash, mucositis, and asthenia [6]. Thus far, conservative therapies such as a VP shunt, seizure control, rehabilitation, and laser therapy have been the only available treatments for MCAP. However, it may be possible in the future to treat MCAP using molecular-targeted therapies.
The underlying PIK3CA-related disorder can lead to cancer development [16]. However, since PIK3CA-related disorders are caused by somatic mosaics and gain-of-function mutations, cancer incidence in these cases is less frequent than in other cancerprone syndromes, such as phosphatase and tensin homolog (PTEN) hamartoma tumor syndrome [16]. Nevertheless, Wilms’ tumor, meningioma, leukemia, and subcutaneous lipoma have been reported in patients with MCAP [5,7]. Therefore, clinicians should regularly follow-up for cancer development, which further highlights the need for early genetic diagnosis.
In conclusion, we report a patient diagnosed in the neonatal period with MCAP and the PIK3CA gene mutation. Recently, therapies targeting the PI3K/AKT/mTOR signaling pathway have emerged as new treatments for PIK3CA-related disorders. Therefore, given that MCAP is a PIK3CA-related disorder, genetic diagnosis is crucial, especially because not all cases of MCAP have a clear clinical presentation. Additionally, because MCAP is a progressive disorder that can potentially lead to cancer development, early diagnosis is essential. Any newborn with macrocephaly, capillary malformation, or distal limb anomalies should undergo brain imaging and genetic evaluation to consider MCAP.
Go to :
 XML Download
XML Download