

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The prevalence of diabetes has risen relentlessly and predisposes an approximate 350 million individuals to various types of devastating complications [1,2]. Mitochondria are organelles with central roles in regulating cellular metabolism and death. Thus, mitochondrial dysfunction, which includes genetic alterations in mitochondrial DNA (mtDNA) and dysregulated mitochondrial-related activities, could lead to a vast array of health concerns, such as diabetes [3]. First, genetic alterations in mtDNA, including mtDNA point mutations, diabetes-related mtDNA polymorphisms, ethnicity-specific mtDNA haplogroups, and low mtDNA copy number, have been reported to be associated with defective insulin secretion, insulin resistance, or both [4-8]. Second, dysregulated mitochondrial activities such as mitochondrial fusion/fission, mitochondrial oxidative stress, and decreased nicotinamide adenine dinucleotide (NAD+) levels correlate with the increased risk of diabetes [9-13]. Considering these pathogenetic links between diabetes and mitochondria, it is plausible that peptides derived from mtDNA play a crucial role in the pathogenesis of diabetes.
Go to : 
MITOCHONDRIAL-ENCODED PEPTIDES
There are currently nine published mitochondrial-encoded peptides: humanin (HN), mitochondrial open reading frame of the 12S ribosomal RNA type-c (MOTS-c), short humanin like peptides (SHLPs) 1-6, and small human mitochondrial open reading frame over serine tRNA (SHMOOSE), which are composed of less than 58 amino acids [14-18]. These microproteins are found intracellularly or in circulation and target various tissues, including skeletal muscle, liver, brain, and immune cells [14,15,17,18]. MOTS-c is composed of 16 amino acids and has an α-helical structure [14], which is the most common secondary structure in naturally-occurring proteins [19-21]. This simple structure of MOTS-c imbues great advantages as a drug. In fact, the α-helices appear to mediate protein folding and protein-protein interactions that are relevant to various physiological functions and diseases [22]. In addition to its structural advantage, MOTS-c interact with and regulate many different nuclear genome-encoded proteins such as mechanistic target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK). These two kinases are responsible for regulating metabolic adaptability and age-related diseases. MOTS-c controls these two kinases and can prevent both diet-induced and T-cell-induced diabetes in mouse models [14,15]. Here, we will focus on the therapeutic potential of MOTS-c in diabetes and age-related diseases.
Go to :
TYPE 1 DIABETES MELLITUS AND MOTS-c
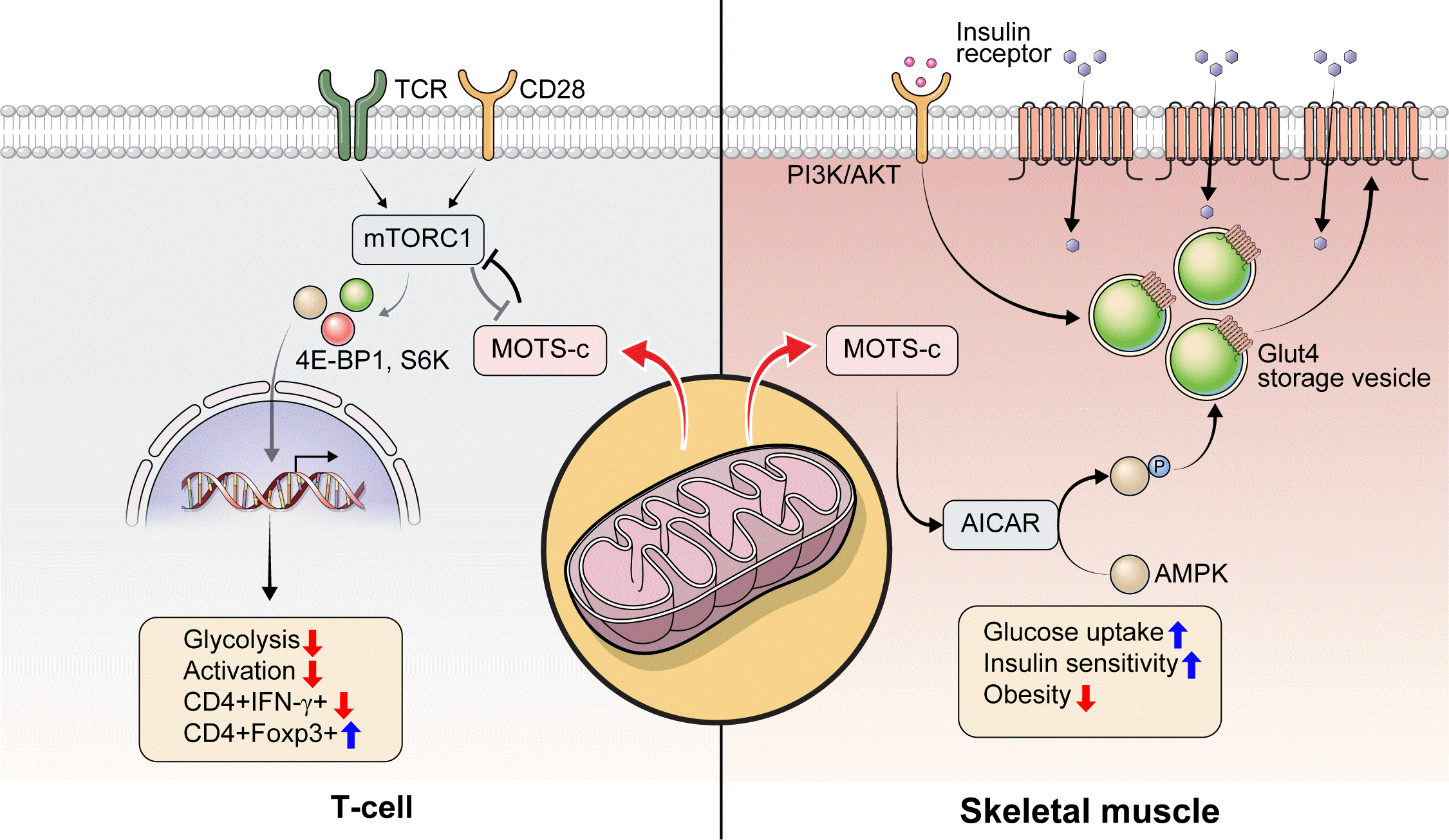
Type 1 diabetes mellitus (T1DM) is a T-cell-mediated autoimmune disease characterized by β-cell destruction [2]. Pancreatic infiltration of autoreactive T-cells (CD4+, CD8+ T-cells) originated from lymphoid organs are thought to be responsible for the β-cell destruction (Fig. 1) [23]. Emerging evidence suggests that mitochondria play a key role in the pathogenesis of autoimmune disease, including T1DM [3]. It is reported that alterations in mitochondrial electron transport [24], mitochondrial reactive oxygen species (mtROS) [25], mitochondrial nitric oxide (mtNO) [26,27], and mitochondrial hyperpolarization [28] of β-cells or immune cells (i.e., T cells) can lead to the pathogenesis of T1DM. These alterations in mitochondrial function and T cell-induced T1DM pathogenesis facilitate the activation and recruitment of T-cells to destroy insulin-secreting pancreatic β-cells. Together, these observations indicate a link between mitochondria and diabetes.
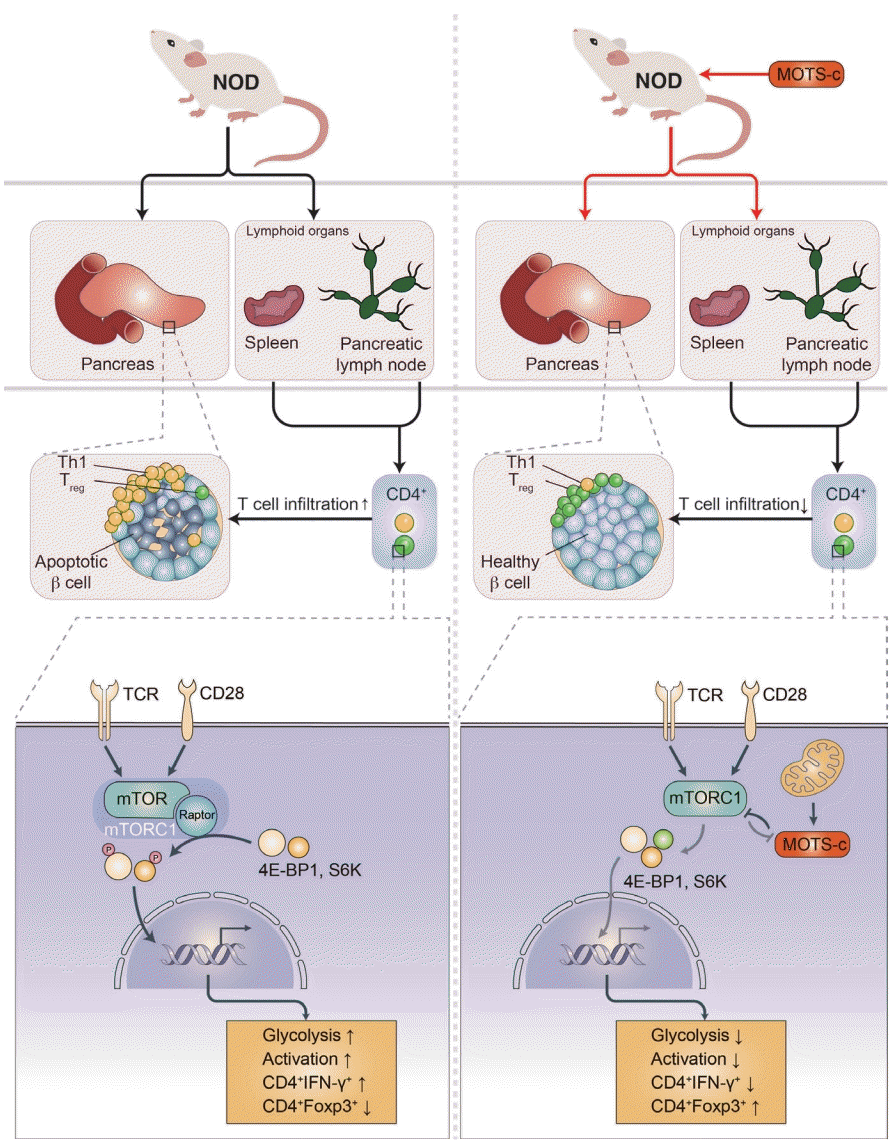 | Fig. 1.Mitochondrial open reading frame of the 12S ribosomal RNA type-c (MOTS-c) prevents type 1 diabetes mellitus (T1DM). T1DM is an autoimmune disease. Activated autoreactive T-cells such as Th1 cells from secondary lymphoid organs (i.e., spleen or pancreatic lymph nodes) infiltrate into the pancreas and cause β-cell destruction. MOTS-c regulates T-cell differentiation and activation (e.g., upregulation of Treg cells) in an mechanistic target of rapamycin complex 1 (mTORC1)-dependent manner and prevents pancreatic infiltration of autoreactive T-cells in non-obese diabetic (NOD) mice. MOTS-c inhibits mTORC1 to lower glycolysis, which influences T-cell differentiation and activation. Inhibition of mTORC1 results in lowered glycolysis, lowered interleukin gamma (IFN-γ), and increased forkhead box P3 (Foxp3) expression level. As a result, MOTS-c treatment in T-cells lowers glycolysis to favor Foxp3+ Treg differentiation and lowers T-cell activation in NOD mice and T1DM patients. TCR, T-cell receptor; 4E-BP1, 4E binding protein 1; S6K, S6 kinase.
|
The role of HN and MOTS-c in T1DM has been investigated using the non-obese diabetic (NOD) mouse model of the disease. HN treatment (n=10/group, 2.0 mg/kg/day; intraperitoneal [IP]) in 5-week-old NOD female mice significantly improved glucose homeostasis, with 70% being normoglycemic with HN treatment compared to 40% in the control group [29]. HN treatment decreased cytokine (tumor necrosis factor alpha, interleukin gamma [IFN-γ])-induced β-cell apoptosis measured by caspase 3/7 activity, improved glucose tolerance measured by intraperitoneal glucose tolerance test (IPGTT), lowered the degree of insulitis, and delayed the onset of diabetes in female NOD mice [29]. As the pathogenesis of T1DM is highly associated with immune cell infiltration, it is possible that HN could regulate immune cell proliferation and function. However, it was unknown whether mitochondrial-encoded peptide treatment regulates proinflammatory immune cells to prevent autoimmune disease, such as T1DM. To answer this question, we assessed the immunoregulatory role of another mitochondrial-encoded peptide, MOTS-c, in NOD mice [14]. We administered MOTS-c (n=28/group, 0.5 mg/kg/day; IP) in 7-week-old female NOD mice until 18 or 30 weeks of age. MOTS-c treatment delayed the onset of autoimmune diabetes in both 18-and 30-week-old NOD mice [14]. MOTS-c improved glucose tolerance and insulin secretion measured by IPGTT and serum insulin, respectively. Also, the treatment of MOTS-c protected pancreatic β-cells against proinflammatory CD4+ IFN-γ+ (Th1 cells) and cytotoxic CD8+ T-cells by lowering the augmentation of T-cells around the pancreatic islets (Fig. 1) [14]. Protein modeling predicted the α-helical domain of MOTS-c to bind Raptor through side chain-side chain interaction. The binding of MOTS-c to Raptor may act as a competitive inhibitor that prevents the binding of TOR signaling (TOS) motifs of 4E binding protein 1 (4E-BP1), S6 kinase (S6K), and proline-rich AKT substrate (PRAS) to Raptor. Consistently, MOTS-c inhibited mTORC1 activation and decreased T-cell glycolysis, Th1 differentiation, and T-cell activation. Furthermore, MOTS-c regulated IFNG and forkhead box P3 (FOXP3) expression in spleen-derived T-cells from C57BL/6J and Jurkat cells. MOTS-c mutants that lack the hydrophobic core (8YIFY11 to 8AAAA11) diminished its inhibitory effect on mTORC1 signaling and IFNG FOXP3 expression. Adoptive transfer of splenocytes from MOTS-c-treated NOD-severe combined immunodeficiency (SCID) mice did not induce diabetes in the immunocompromised NOD-SCID mice, in part, by increasing the proportion of pancreatic Foxp3+ Treg cells (Fig. 1) [14]. Furthermore, treatment of MOTS-c prevented the activation of human T1DM patients derived T cells [14]. Together, we unveiled that MOTS-c has a robust immunoregulatory function in autoimmune T1DM.
Two human T1DM studies suggest a potential link between T1DM pathogenesis and circulating mitochondrial-encoded peptides levels. One study enrolled 21 to 25 year old men and women who were T1DM patients (n=41) or healthy (n=21) to measure HN levels in serum. Circulating HN levels of men with T1DM (1,016±139 pg/mL, n=23) were higher compared to those of healthy men (762±154 pg/mL, n=10) [30]. In line with previous reports, we compared serum MOTS-c levels in human subjects with and without T1DM. Unlike HN, serum MOTS-c levels were lower in T1DM patients compared to healthy controls [14]. Overall, these data suggest a connection between circulating mitochondrial-encoded peptides and the pathogenesis of autoimmune diabetes.
Go to :
TYPE 2 DIABETES MELLITUS AND MOTS-c
Type 2 diabetes mellitus (T2DM) is characterized by the dysregulation of multiple metabolic pathways involved in impaired insulin secretion, insulin resistance, or a combination of both [1]. Both impaired insulin secretion and insulin resistance may result from mitochondrial dysfunction, high production of reactive oxygen species, and low levels of adenosine triphosphate [31,32]. Over the past 30 years, numerous studies have examined the relationship between mitochondria, mtDNA, and T2DM. In a population-based prospective cohort study, newly diagnosed T2DM patients had 25% lower mtDNA copy number in peripheral blood mononuclear cells (PBMCs) compared to a non-diabetic group (102.8±41.5 vs. 137.8±67.7 copies/pg template DNA) [33]. Furthermore, decreased mtDNA copy number correlated with waist-hip circumference ratio, fasting hyperglycemia, and high blood pressure [33]. In another population-based cohort study, the mtDNA copy number in peripheral blood cells of offspring of diabetic or non-diabetic patients were compared. All subjects showed normoglycemia, but the mtDNA copy number of PBMCs was decreased in the offspring of diabetic patients compared to those of non-diabetic healthy controls [34]. Similar findings were also observed in healthy young men, whereby the PBMC mtDNA copy number correlated with insulin sensitivity, insulin secretion, and fat oxidation rate [35,36], consistent with other reports from many other groups [3,4,17,28,31, 32,37-47]. Taken together, mitochondria and mtDNA derived factors likely play crucial roles in the pathogenesis of T2DM [4,43].
MOTS-c is a mitochondrial-encoded peptide that is found in multiple types of cells [14,15,17,48-57]. Circulating MOTS-c levels have been measured in individuals with T2DM, obesity, and sleep apnea [49,51,52,55-58]. In a cross-sectional study, 225 subjects (healthy controls, 68; pre-diabetes, 33; T2DM with glycosylated hemoglobin [HbA1c] <7%, 31; and >7%, 93) serum MOTS-c levels were lower in patients with inadequately controlled T2DM (HbA1c >7%) [58]. In another study with 97 subjects (13 obese female children, 27 obese male children; and 17 healthy female and 40 healthy male children, age between 5 and 14 years old), serum MOTS-c levels were significantly lower in the obese group compared with the control group (472.61±22.83 ng/mL vs. 561.64±19.19 ng/mL, P<0.01). There was a difference between non-obese and obese men in terms of the serum MOTS-c concentration, but not in women [52]. These human studies suggest a potential relationship between circulating MOTS-c levels and obesity or T2DM sexual dimorphism. The mechanism of MOTS-c in association with T2DM pathologies has been studied. MOTS-c has been reported to reduce insulin resistance by targeting the skeletal muscle in mice fed a high-fat diet (Fig. 2) [15]. MOTS-c is intertwined with the folate cycle, 5-aminoimi-dazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), and AMPK signaling. It can promote AICAR accumulation, AMPK activation, and the translocation of glucose transporter type 4 (GLUT4) to the plasma membrane in muscle cells (Fig. 2) [15]. Increased expression and membrane localization of GLUT4 facilitates glucose uptake into muscle cells to support adaptive metabolism and prevents hyperinsulinemia [15]. MOTS-c can regulate nuclear genes associated with metabolic stress, such as activating transcription factor 1 (ATF1) and nuclear factor-erythroid factor 2-related factor 2 (NRF2), by translocating to the nucleus and interacting with chromatin [54]. These studies suggest that MOTS-c is a novel metabolic regulator that can be a therapeutic target for T2DM.
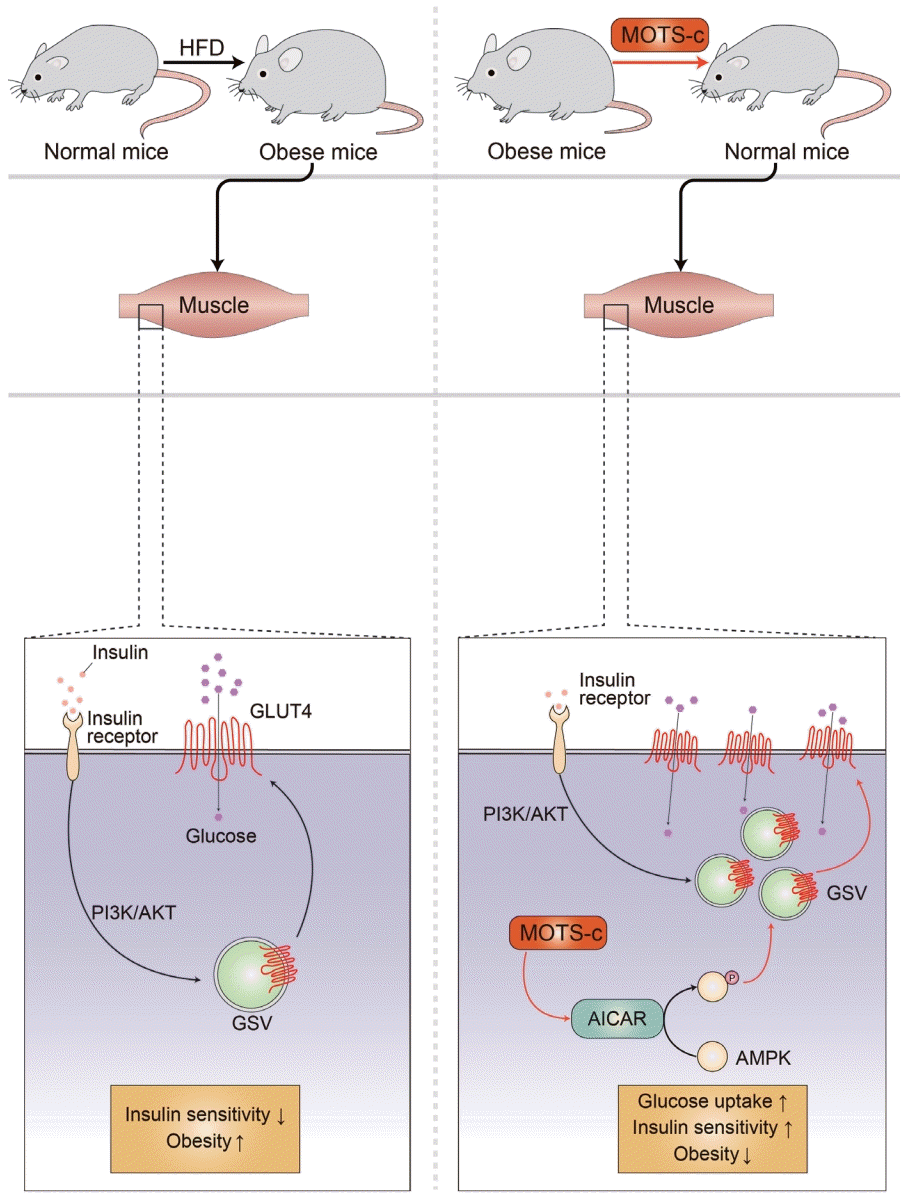 | Fig. 2.Mitochondrial open reading frame of the 12S ribosomal RNA type-c (MOTS-c) reduces insulin resistance in high-fat diet (HFD) induced obese mice. Obesity is associated with increased insulin resistance. MOTS-c targets the skeletal muscle to regulate metabolic homeostasis. MOTS-c increase 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) and activates AMP-activated protein kinase (AMPK). This leads to the upregulation of glucose transporter type 4 (GLUT4) expression level in muscle cells. GLUT4 is a glucose transporter that is responsible for glucose uptake in the skeletal muscle and adipose tissues. MOTS-c treatment in HFD induced obese mice prevents obesity and hyperinsulinemia by regulating GLUT4 in an AMPK-dependent manner. PI3K, phosphoinositide 3-kinase; GSV, GLUT4 storage vesicle.
|
Go to :
AGING AND MOTS-c
Emerging studies increasingly connect the functions of MOTS-c to aging. Fuku et al. [53], showed that Asian centenarians have a different variant of a polymorphism located in the open reading frame of MOTS-c, suggesting a close relationship between the MOTS-c variant and exceptional longevity in humans. Notably, the hydrophobic and cationic domains of MOTS-c (8YIFY11 and 13RKLR16, respectively) have key functional roles. The loss of the hydrophobic domain by substituting the residues with alanine (8YIFY11>8AAAA11) revealed that the hydrophobic domain is required for its nuclear translocation and nuclear gene regulation [54]. This was confirmed in T-cells (Jurkat), whereby the overexpression of MOTS-c increased oxidative phosphorylation (OXPHOS) and decreased glycolysis. However, the overexpression of the MOTS-c mutant that lacked the hydrophobic domain (8AAAA11) did not exhibit the metabolic shift from glycolysis to OXPHOS [14]. Yet, although these studies focused on the nuclear and metabolic roles of MOTS-c, it can be inferred that MOTS-c polymorphisms can have a functional impact, including metabolism and adaptive responses. Other mtDNA polymorphisms also contribute to T2DM risk in both European (m.4216T>C and m.4917A>G variants) and Asian populations (N9a haplogroup) [59,60]. Further studies are needed to evaluate the mechanistic connection between mtDNA polymorphisms and mitochondrial-encoded peptides in T2DM.
One of the major changes that occur during aging is the dysregulation of the immune response that leads to a chronic low-grade inflammatory state, which is thought to be a major risk factor for multiple chronic age-related dysfunctions and/or diseases, including hair loss, neurodegeneration, cancer, osteoporosis, cardiovascular disease, atherosclerosis, sarcopenia, and T2DM (Fig. 3) [15,49,51,52,55,56,61-66]. Interestingly, the discovery of MOTS-c was inspired by a study on inflammatory responses [15]. Tsuzuki et al. [67] reported that transcripts from the mitochondrial ribosomal genes were induced under interferon-stimulated conditions in monocyte-like cells. To test the hypothesis that mitochondrial-encoded peptides are associated with inflammatory processes, we measured several cytokines in NOD mice after MOTS-c treatment. We found that MOTS-c treatment in NOD mice can reduce peri-insulitis in pancreatic islets, regulate cytokines such as interleukin-10 (IL-10) and IFN-γ in serum and isolated splenic T-cells, and prevent T-cell-driven disease transfer to NOD-SCID mice [14]. This study was one of the first to reveal the direct regulation of immune cells by mitochondrial-encoded peptides. Notably, interferon responses and inflammation are hallmarks of cellular senescence and aging [68-70].
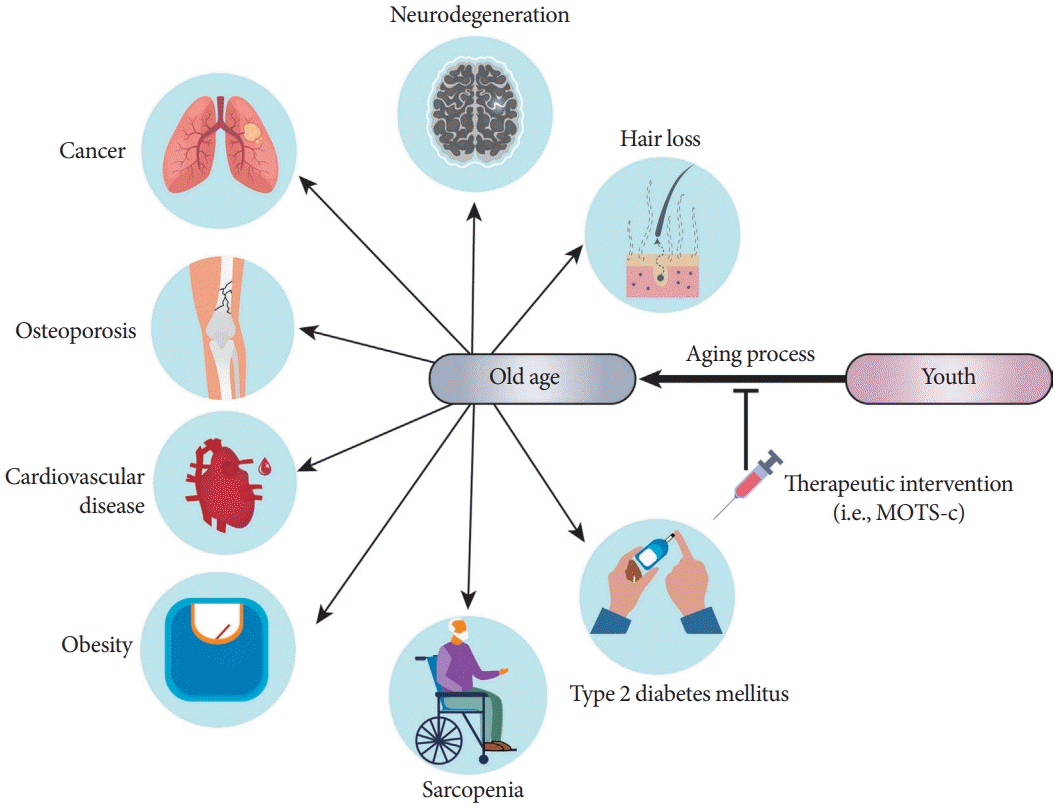 | Fig. 3.Mitochondrial open reading frame of the 12S ribosomal RNA type-c (MOTS-c) is a potential target against aging-related diseases. Aging is associated with number of diseases. Traditionally, drug discovery efforts target each of these diseases individually. A complementary approach is to develop a therapeutic reagent to treat the root of these diseases, dubbed geroscience. MOTS-c is a potential therapeutic target for multiple aging-related diseases, including neurodegeneration, osteoporosis, cardiovascular disease, atherosclerosis, sarcopenia, type 2 diabetes mellitus, and obesity. Future studies are needed to unveil the efficacy and safety of MOTS-c treatment for aging-related diseases.
|
Other studies corroborate the relationship between aging, senescence, and MOTS-c. MOTS-c expression is lower in senescent human fibroblast cells induced by replication stress [71]. In mouse skeletal muscle and circulation, MOTS-c level also declined with age [15]. In human subjects of young (18 to 30 years), middle (45 to 55 years), and older (70 to 81 years) age (n=25/group), the plasma MOTS-c level was reduced with aging (young vs. middle, P<0.01; young vs. older, P<0.001; middle vs. older, P<0.05) [15]. Recent findings showed that 2 weeks of systemic MOTS-c treatment significantly increases the physical capacity of old mice (22 months), allowing them to double their running time on a treadmill and effectively outrun their middle-aged (12 months) counterparts [72]. Notably, in humans, exercise (i.e., stationary bicycle) considerably raises the levels of endogenous MOTS-c in the skeletal muscle and in circulation, indicating interorgan mitochondrial communication [72]; parallel results have been reported in rodents [73,74]. Indeed, exercise also increases endogenous levels of MOTS-c in hypothalamic proopiomelanocortin (POMC) neurons by exercise-related cytokines such as IL-6. MOTS-c stimulates POMC transcription in an IL-6-dependent manner to increase sympathetic nerve activity, which plays a crucial role in exercise [75,76]. Late-life initiated (24 months) intermittent MOTS-c treatment (3× weekly) increased healthy lifespan in mice, suggesting the potential for further development as an anti-aging intervention [72]. Further studies are needed to unveil the mechanisms by which MOTS-c regulates inflammation, cellular senescence, and aging [62].
Go to :
FUTURE THERAPEUTICS AND MOTS-c
The discovery of MOTS-c and other mitochondrial-encoded peptides may pave a new conceptual way for the treatment of metabolic, age-related, and autoimmune diseases. Pre-clinical data on MOTS-c holds much therapeutic potential as they demonstrate effective prevention/protection against aging and a broad range of age-related dysfunction and diseases. Several groups have reported the beneficial role of MOTS-c treatment using various animal models, including ovariectomy-induced metabolic dysfunction [63] and ovariectomy-induced bone loss (Fig. 3) [64]. Clinical trials to test the therapeutic potential of MOTS-c is ongoing and currently limited, including a clinical trial using a MOTS-c analog for fatty liver and obesity (clinical trial #NCT03998514). Notably, we previously showed that in human 143B-osteosarcoma-origin cybrid cells harboring the mtDNA 3243A>G mutation Rho0 cells, MOTS-c expression was decreased. In the cybrid cells, treatment or overexpression of MOTS-c was able to increase mitochondrial complex subunits (ubiquinol-cytochrome c reductase, complex III; and NADH:ubiquinone oxidoreductase subunit A1, complex I) mRNA expression levels, but it did not show metabolic effects [48]. This suggest that MOTS-c may have an impact on mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome caused by the mtDNA 3243A>G mutation in tRNAleu, but the underlying functional connection still remains unknown.
MOTS-c is the first mitochondrial-encoded peptide that has been subjected to clinical trials, uncovering the mitochondrial genome as a source of therapeutics and drug targets. Peptides are vital physiological mediators that are attractive therapeutic candidates with their high potency, specificity, and low toxicity. Nonetheless, there is still much to understand about MOTS-c and other mitochondrial-encoded peptides, including their basic molecular mechanisms, stability in biological systems, oral bioavailability, and relevance to a broad range of diseases and conditions.
Go to :
 XML Download
XML Download