

 PDF
PDF Citation
Citation Print
Print



Introduction
Early hormonal treatment of congenital hypothyroidism (CH) can prevent mental retardation. The frequent etiology of CH is thyroid dysgenesis, which includes agenesis, ectopy, and hypoplasia, followed by defects in thyroid hormone biosynthesis (dyshormo-nogenesis).1) Reduced thyroxine synthesis during dyshormonogenesis induces increased thyrotropin (TSH) secretion through reduced negative feedback of thyroxine. Consequently, affected patients has congenital goiter, otherwise postnatal goiter without L-thyroxine (LT4) supplement.
Even if CH is diagnosed in the newborn period based on the levels of TSH and free T4, etiologic diagnosis is not required before starting LT4 replacement because the management remains the same, irrespective of the cause. At the age of 3 years, reevaluation of CH patients based on hormone levels and thyroid imaging, should be performed after stopping LT4 for 4 to 6 weeks.2) Scintigraphy is useful for the diagnosis of ectopic thyroid. For the diagnosis of TPO deficiency, the perchlorate discharge test is useful,3) but is not commonly used due to the risk of radiation exposure. In addition, a genetic diagnosis of CH is not essential for determining the treatment because treatment is always LT4 replacement. However, molecular diagnosis of these heterogeneous entities could provide prognostic insights and reliable genetic counseling along with more specific targets for molecular therapies.4) Panel sequencing based on next-generation sequencing (NGS) is useful in cases of dyshormonogenesis.5)
In patients with dyshormonogenesis, the thyroid peroxidase (TPO) gene is the most commonly mutated.6) However, recent studies from China, Thailand, and Korea have reported that dyshormonogenesis is more common than dysgenesis, and dual oxidase2 (DUOX2) is the most commonly mutated gene.7-9) Multinodular goiters have also been reported to exhibit dyshormonogensis.10) Herein, we report the sequential changes in the ultrasonographic appearance of a solitary thyroid nodule during LT4 supplementation in a patient who was genetically confirmed to have a TPO mutation using NGS.
Go to : 
Case Report
An 8.2-year-old girl visited our clinic because of thyroid enlargement. In her medical history, neck swelling was noted during the newborn period, but further investigation was not performed because of a normal newborn screening test. Upon examination, her vital signs were nonspecific. Thyroid function test was normal as follows: free T4 0.91 ng/dL (normal, 0.7-1.48), TSH 4.68 uIU/mL (normal, 0.35-4.94) (Table 1). Thyroid autoantibodies were not detected. 99mTc scan showed a diffusely enlarged thyroid gland with increased uptake, and ultrasonography (US) showed diffuse thyroid enlargement with heterogeneous echogenicity (Fig. 1). LT4 was administered as dyshormonogenetic CH was suspected. The goiter decreased in size; however, it did not disappear completely. At 11.5 years of age, the goiter size increased again and thyroid function became mildly hypothyroid after discontinuation of LT4 for 6 months (Table 1). US showed diffuse enlargement of the thyroid gland with heterogeneously decreased echogenicity, as well as an isoechoic solid nodule of size 1.8×1.7×1.6 cm with a peripheral halo in the inferior aspect of the right thyroid. The nodule was reported to be a benign follicular lesion with nodular hyperplasia on fine-needle aspiration biopsy (FNAB) (Fig. 2). After resuming LT4, the goiter size decreased again, and the euthyroid state was maintained until the patient stopped taking medication by her own accord at 13.5 year of age. Thereafter, the goiter size changed according to the functional status of the gland. At 18 years of age, the nodule size was 1.1×0.9 cm in the same location. At 22 years of age, peripheral calcification appeared around the isoechoic solid nodule previously reported, without a change in the size (Fig. 3). The radiologist reported category 3 malignancy risk of the nodule according to the Korean Thyroid Imaging Reporting and Data System (K-TIRADS). NGS panel for CH reported two variants of the TPO gene and one variant of the dual oxidase maturation factor (DUOXA2) gene. The first variant of TPO, c.[2757del](p.[Met921Trpfs*53]) was a pathologic variant from the asymptomatic father, while the second variant, c.[1580G>T](p.[Trp527Leu]) was likely pathogenic variant from the asymptomatic mother. The heterozygote variant of the DUOXA2 gene with c.[535T>C](p.[Tyr179His]) is value of uncertain significance. The patient is scheduled to undergo regular US to monitor the progression of the thyroid nodule.
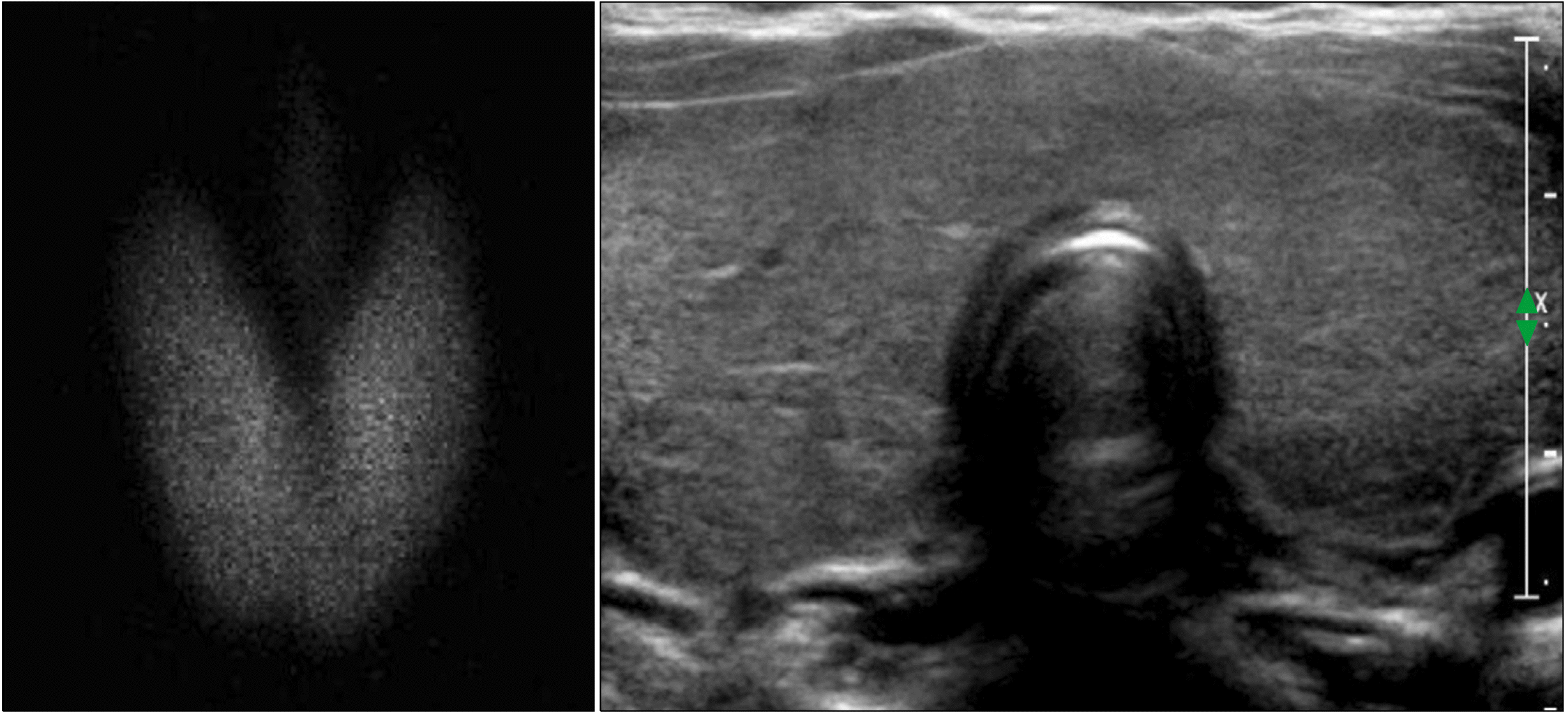 | Fig. 1Initial thyroid examination at 8.2 years of age, shows diffuse enlargement with increased up-take in the 99mTc scan (left) and diffuse thyroid enlargement with heterogeneous echogenicity in ultrasonography (right).
|
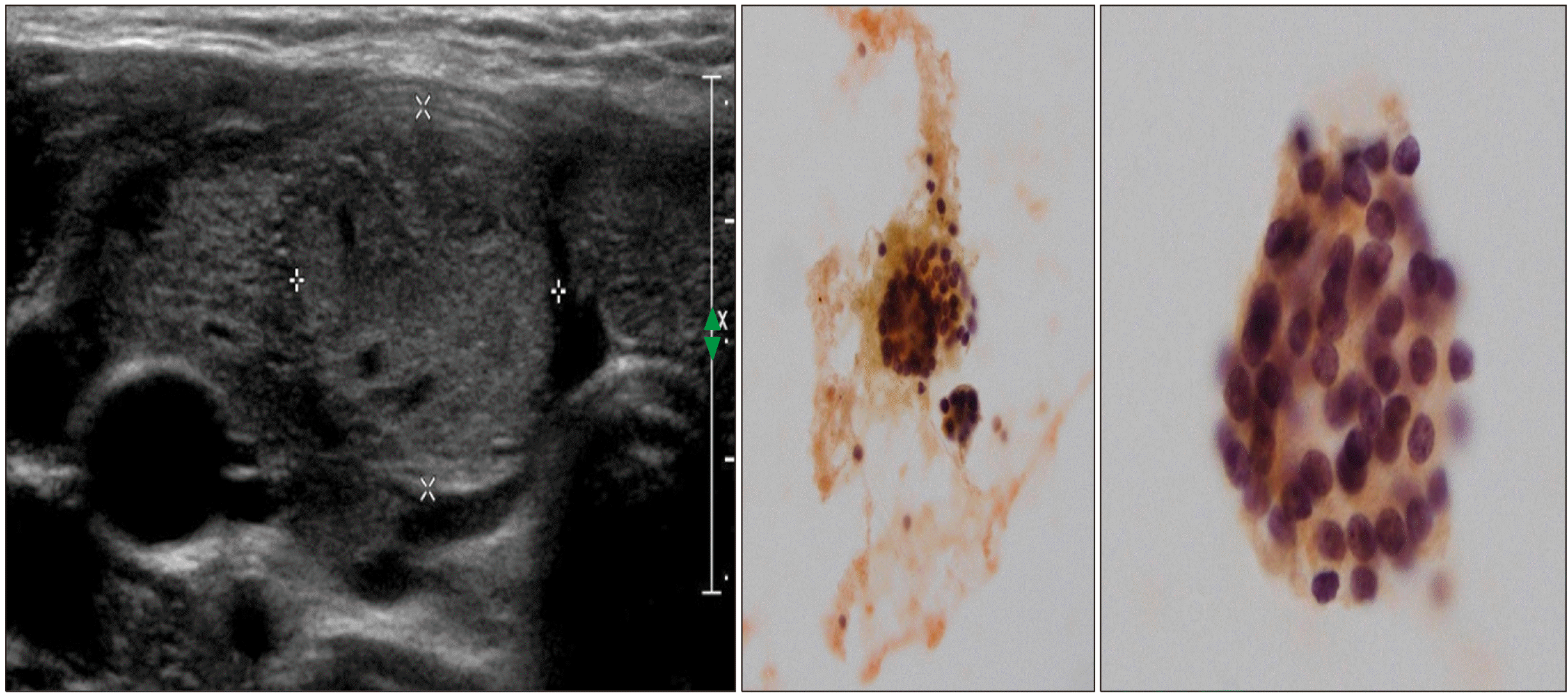 | Fig. 2At 11.6 years of age, an isoechoic solid nodule with a peripheral halo is visible in the right thyroid gland with diffusely enlarged and heterogeneously decreased echogenicity of the entire gland (left). Aspiration biopsy shows a benign follicular lesion favoring nodular hyperplasia (middle and right).
|
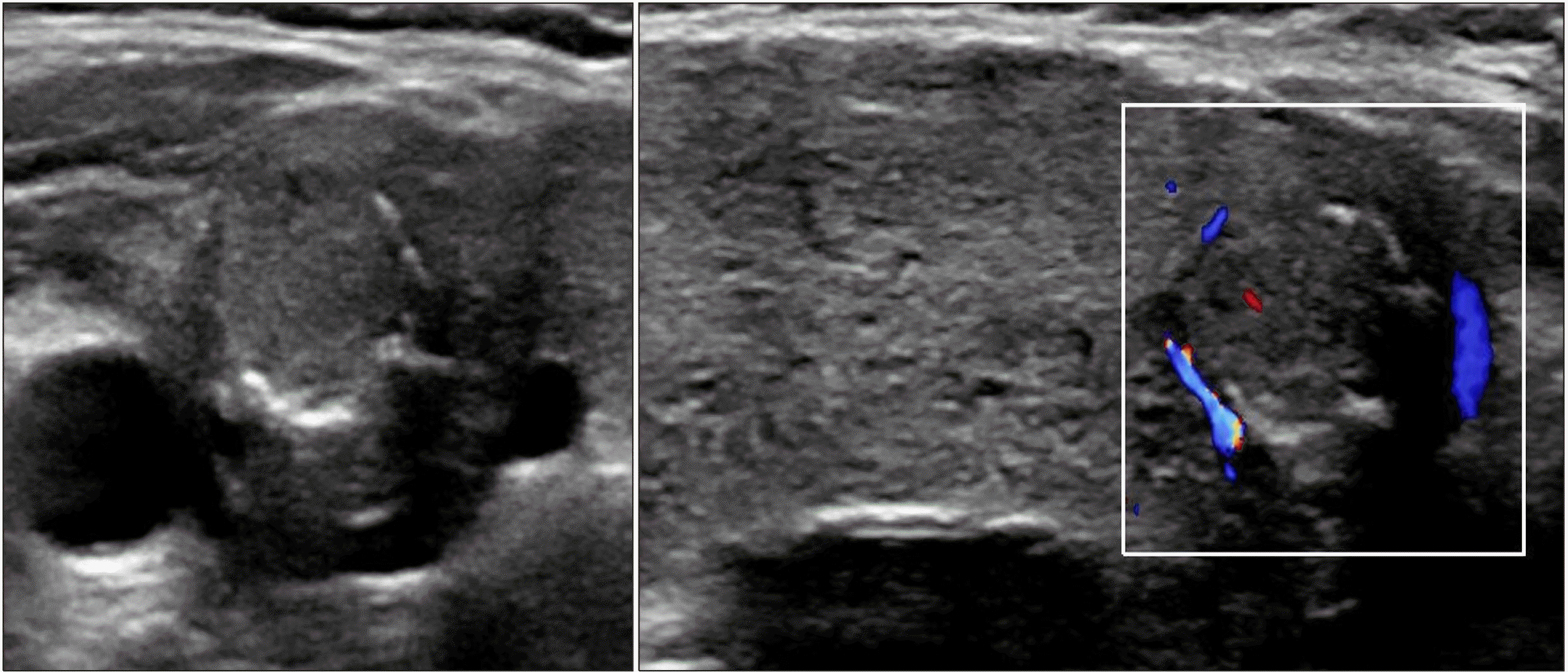 | Fig. 3At 22 years of age, a well-defined isoechoic solid nodule with peripheral calcification in the right inferior thyroid can be seen, along with an enlarged thyroid gland with diffuse heterogenous echogenicity and increased vascularity.
|
Table 1
Laboratory findings and clinical course
![]()
Go to :
Discussion
Thyroid hormone biosynthesis involves the following stages: iodide transport, thyroglobulin synthesis, oxidation and organification, H2O2 generation, coupling of iodinated tyrosines within thyroglobulin, endocytosis of colloids and hormone release, and recycling of iodide. Defects in biosynthetic pathway cause approximately 10-15% cases of CH. Seven genes are known to cause dyshormonogenesis.5)
The most common mutated gene in thyroid dyshormonogenesis is TPO gene. A study from Slovenia, Bosnia, and Slovakia reported that up to 46% of thyroid dyshormonogenetic participants had a TPO gene mutation.11) Since the first report of a biallelic mutation of TPO in 1992 (6), 188 (professional ver.) different TPO mutations have been identified in HGMDⓇ Professional 2021.4 (http://www.hgmd.cf.ac.uk). Most patients with TPO mutations have congenital goiter. They show profound hypothyroid state, high uptake of radioactive iodine on scintigraphy, elevated thyroglobulin levels, and positive perchlorate discharge test.5) However, normal development without goiter was reported in a sibling who received LT4 immediately after birth compared to two other goitrous siblings who received LT4 from 6 months of age.3) Our patient also showed diffuse goiter with increased uptake on 99mTc scintigraphy and elevated thyroglobulin levels. Unfor-tunately, the perchlorate discharge test was not carried out because of the risk of radiation exposure.
TPO mutated CH has autosomal recessive pattern of inheritance, although monoallelic TPO defect comprises approximately 20%. Maybe unconfirmed cryptic mutations in introns or regulatory regions can explain this phenomenon.1) The NGS panel for CH in our case revealed three relevant variants, two in TPO and one in DUOXA2. The first TPO variant, c.[2757del] (p.[Met921Trpfs*53]), inherited from an asymptomatic father, is a well-known pathogenic frameshift variant that is associated with a stop codon. The incidence of the second TPO variant, c.[1580G>T](p.[Trp527Leu]), inherited from an asymptomatic mother, is 0.0058% in the population database (gnomAD); this has been reported as likely pathogenic. The incidence of the variant of the DUOXA2 gene, c.[1580G>T](p.[Trp527Leu]), has been reported to be 0.056% in the population database (gnomAD) or 0.14% (KRGDB). This variant is also classified as value of uncertain significance but is predicted to be deleterious in silico (SIFT, Polyphen2, MutationTaster), and has been suspected to be related to the disease.
Some investigators have suggested that DNA collection is necessary for future mutational analysis if dyshormonogenetic goiter is suspected in a patient based on the identification of the thyroid gland in US. Genetic counseling, differentiating between transient and permanent forms of CH, and predicting iodine supplementation in iodotyrosine deiodinase (IYD) gene defects are possible after molecular diagnosis in patients with dyshormonogenesis.1) However, the standard treatment remains LT4 replacement.
In a clinicopathological study of 56 cases, 75% of dyshormonogenetic goiters occurred before the age of 24 years. The thyroid glands were enlarged, multinodular, and cellular nodules with solid and/or microfollicular patterns were the most common microscopic alterations. In 18% of cases, the degree of hyperplasia and atypia led to a misdiagnosis of follicular, papillary, or undifferentiated carcinoma.10) Some investigators have emphasized regular tracing and LT4 medication to prevent thyroid stimulation from elevated TSH.4) Our patient exhibited diffuse goiter with normal thyroid function without autoantibodies at the age of 8 years. Unfortunately, normal neonatal screening test results prevented further investigations in this case. Goiter decreased after 3.5 years of follow-up with LT4 medication. However, the goiter aggravated and thyroid function slightly deteriorated after 6 months’ off treatment. A diffusely enlarged thyroid gland with solid nodules was observed via US. The pathological diagnosis of the aspirated specimen was a benign follicular lesion favoring nodular hyperplasia rather than papillary carcinoma, although it was non-typical.
As childhood thyroid nodules are more often malignant (up to 25%), a more aggressive management approach is justified. Cases of thyroid cancer have been reported in CH due to dyshormonogenesis, usually with follicular histology.12) Several authors have reported that multinodular goiters associated with dyshormonogenesis can be malignant or proceed to real malignancy. Minimally invasive follicular thyroid carcinoma was reported in TPO mutated patient with large multinodular goiter during a 29-year follow-up. However, genetic or environmental factors may have been implicated in this case because the serum TSH levels were not elevated.13) The largest data from 20 TPO-deficient patients followed-up for 43 years showed that 61% developed multinodular goiter, thyroidectomy was performed in 24%, and invasive follicular thyroid carcinoma was found in one case. Moreover, elevated lifetime TSH levels was not associated with goiter occurrence.14) A biallelic thyroglobulin (Tg) mutation was reported in two siblings aged 25 and 31 years. They had similar histories of LT4 medication since childhood, subtotal thyroidectomy for pressure symptoms, and recurrent goitrous hypothyroidism. The older sibling developed a metastasis from microscopic FVPTC after 8 years. In contrast, the younger sibling had a benign dyshormonogenetic goiter on FNAB. The older sibling was frequently noncompliant with LT4. Therefore, malignant transformation of follicular cells by prolonged TSH stimulation was suggested.15) Another frameshift TPO mutated case with complex pathology underwent thyroidectomy.16) If potential neoplastic changes are truly related to atypical nucleus in dyshormonogenesis, rapid genetic diagnosis would be beneficial for patients with dyshormonogenesis.
TPO employs H2O2 as an oxidizing agent for iodine. Timely and spatially regulated activity of NADPH oxidase enzymes (NOX/DUOX) can produce H2O2, a reactive oxygen species (ROS), for various physiologic actions. On the other hand, improper release of ROS from NOX4 is associated with pathologic thyroid tumorigenesis.17) Therefore, the suppression of TSH may be justified with sufficient LT4 to reduce oxidative stress.
Until now, no clear causal relationship has been identified between CH and thyroid cancer. The most widely known causative genes for thyroid cancer (ex. RET, BRAF, RAS, TP53 etc.) and the most detected genes for CH (ex. DUOX2, TSHR, DUOXA2, TPO, SLC26A4 etc.) are very different, in some reports, TPO, TG, and TSHR mutations that are common in CH have been found in the tissues of thyroid cancer. But, it is difficult to identify whether the cause of malignancy is directly due to genetic mutation or the chronic increase of TSH and high oxidative stress in CH.18) Traditional risk factors of thyroid cancer in children are known to be iodine deficiency, autoimmune thyroid disease, and radiation exposure until now. Therefore, somatic mutation and epigenetics contribute much more to the occurrence of malignancy in CH than genetic mutations themselves, because the genes responsible for thyroid cancer are very different from those responsible for CH.
According to the established guidelines for thyroid nodules in pediatric patients, several sonographic features are helpful for selecting patients for FNAB evaluation. Microcalcification, lymph node changes, growing nodule size with LT4 medication, and increased intranodular vascularization on the color Doppler are the sonographic features associated with the increased possibility of malignancy.12) Other sonographic findings of higher malignancy risk in pediatric thyroid nodules included taller-than-wide shaped large solid parenchyma with speckled calcification and absence of smooth margin.19)
In our case, a solid nodule was detected at 11.5 years of age, after 3.5 years of LT4 treatment under the impression of dyshormonogenetic CH, and peripheral calcification around the isoechoic nodule appeared another 10 years later without change in the nodule size. FNAB was not performed during the last follow-up US at 22 years of age because the nodule size and calcification pattern had not changed. The indication for FNAB in cases of category 3 is a nodule size of more than 1.5 cm in K-TIRADS.20) However, frequent follow-up monitoring of the nodule over time is necessary.
In conclusion, early molecular diagnosis and sufficient TSH suppression are important management in childhood dyshormonogenesis to prevent thyroid nodule formation. As with the currently recommended guideline, it would be sufficient to perform US every two to three years and FNAB if necessary in patients with CH (especially dyshormonogenetic goiter).
Go to :
 XML Download
XML Download