

 PDF
PDF Citation
Citation Print
Print


Introduction
Brief overview
Mechanisms of base editing
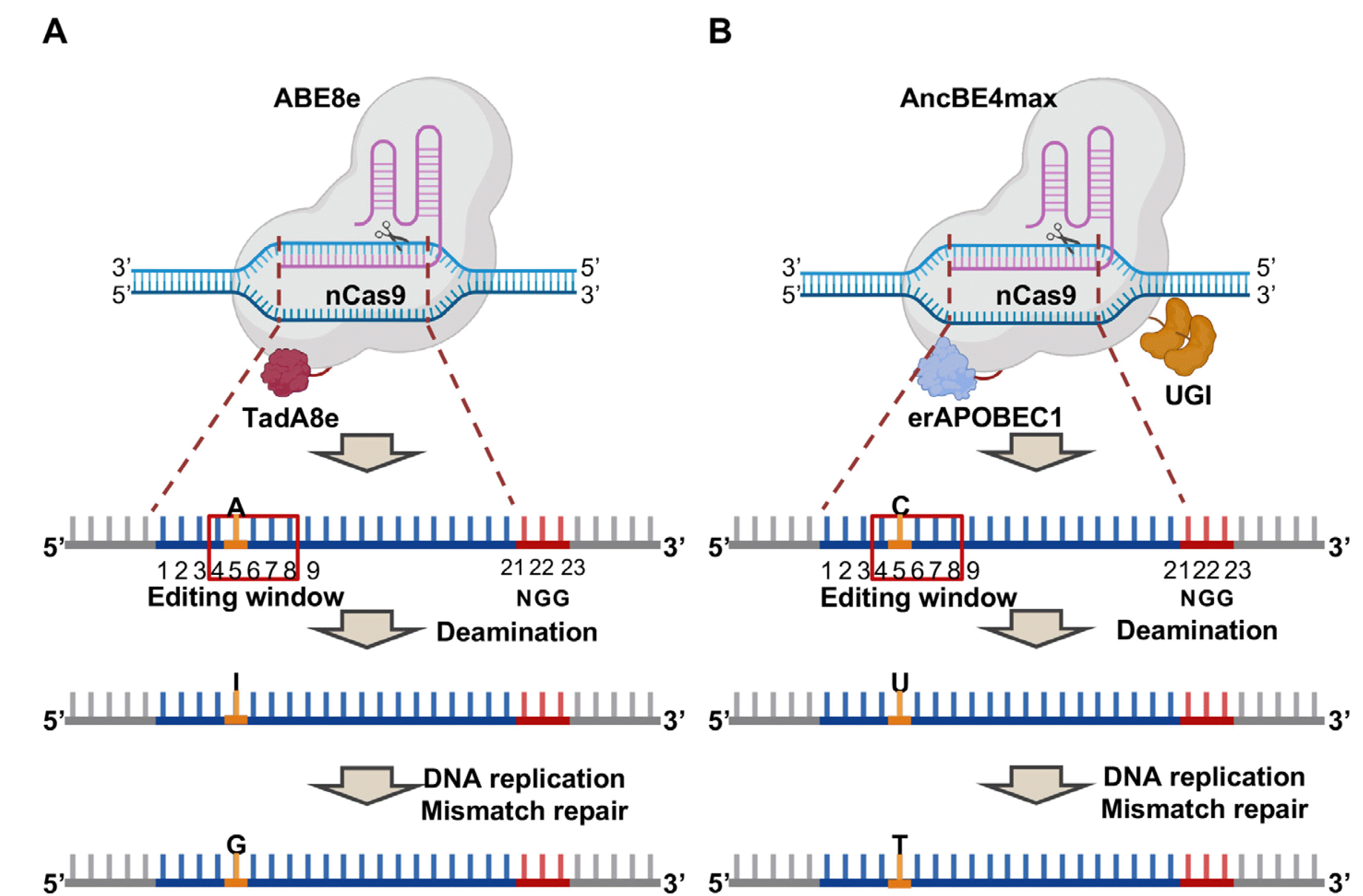 | Fig. 1Composition and mechanisms of ABE8e and AncBE4max. Graphical scheme for composition and mechanisms of (A) ABE8e and (B) AncBE4max, Red box indicates editing window, bases, colored in blue indicates spacer sequence, PAM sequence is colored in red, and the target base for each base editor is colored in yellow. Number indicates the position in the spacer sequence. A for Adenine, I for Inosine, G for Guanine, C for Cytosine, U for Uracil land T for Thymine. Created with BioRender.com.
|
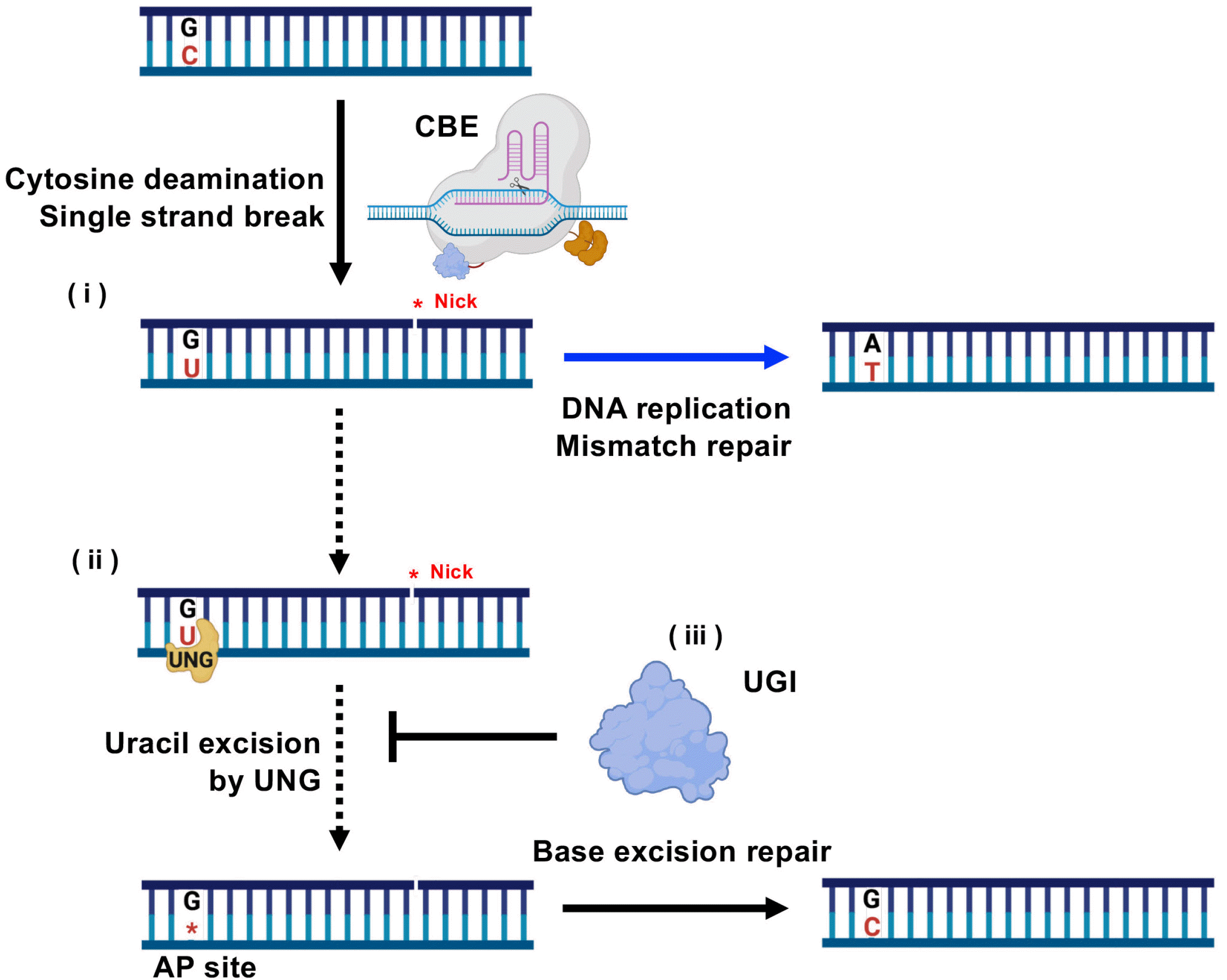 | Fig. 2Role of DNA replication, mismatch repair, base excision repair and UNG in C to T conversion. (i) Nickase activity of nCas9 in CBE induces nick on the editing strand and deaminase activity in CBE produces G:U mismatch. G:U mismatch is converted to A:T via DNA replication followed by mismatch repair. (ii) Alternatively, G:U mismatch, recognized by base excision repair (BER), is removed by UNG to produce AP site, forming G:C. (iii) UNG activity to impair the G-to-A substitution is the co-expression of UNG inhibitor (UGI). Created with BioRender.com.
|
Prerequisites for base editing
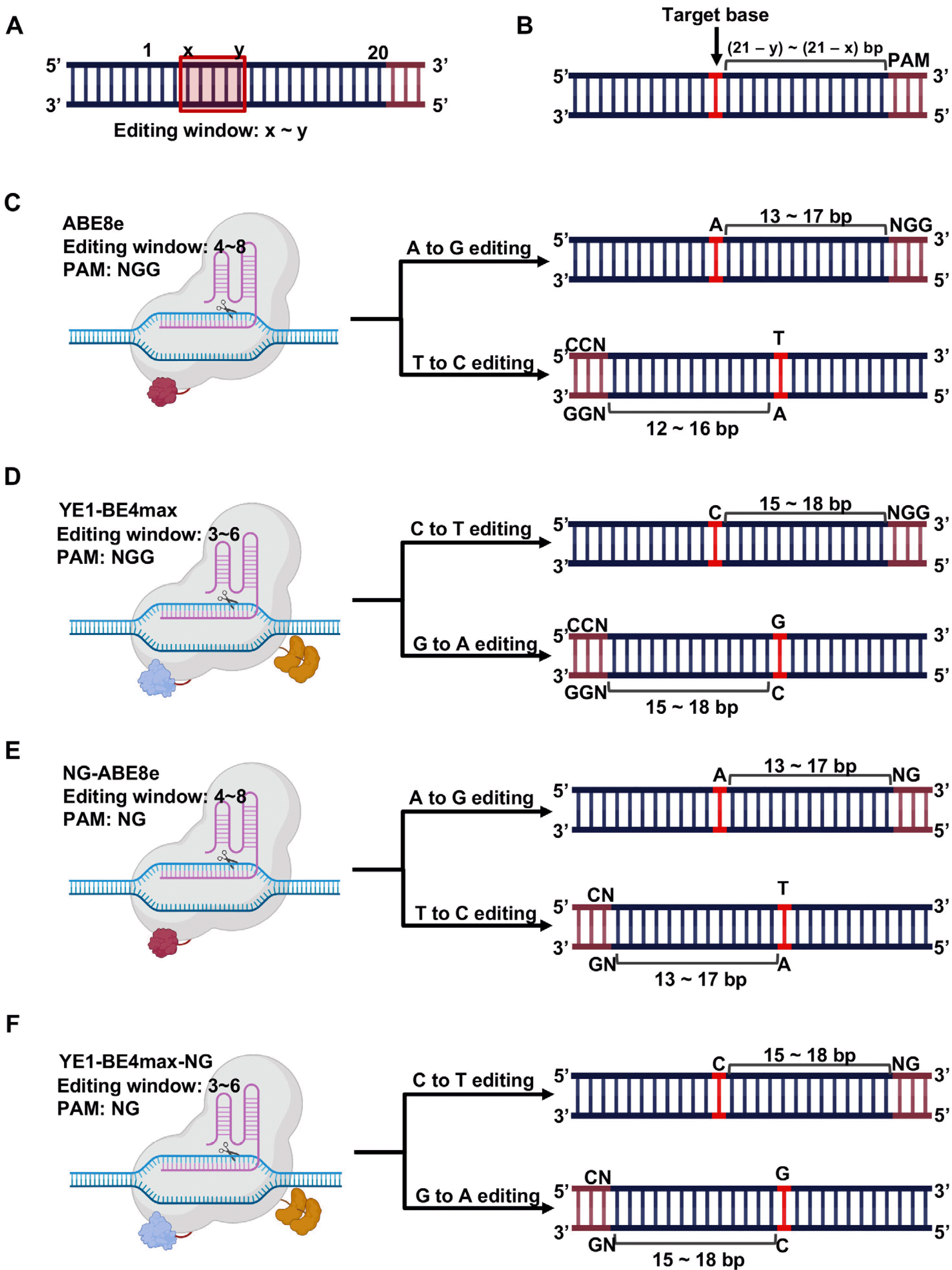 | Fig. 3PAM requirement for base editing. (A) Graphical scheme for editing window of BEs, Red box indicates editing window. ‘x’ and ‘y’ indicates start and end position of editing window respectively. (B) Target base is colored in red and PAM sequence is colored in brown. Graphical scheme of PAM requirement for, (C) ABE8e, (D) YE1-BE4max, (E) NG-ABE8e, and (F) YE1-BE4max-NG. Target base is colored in red and PAM sequence is colored in brown. Created with BioRender.com.
|
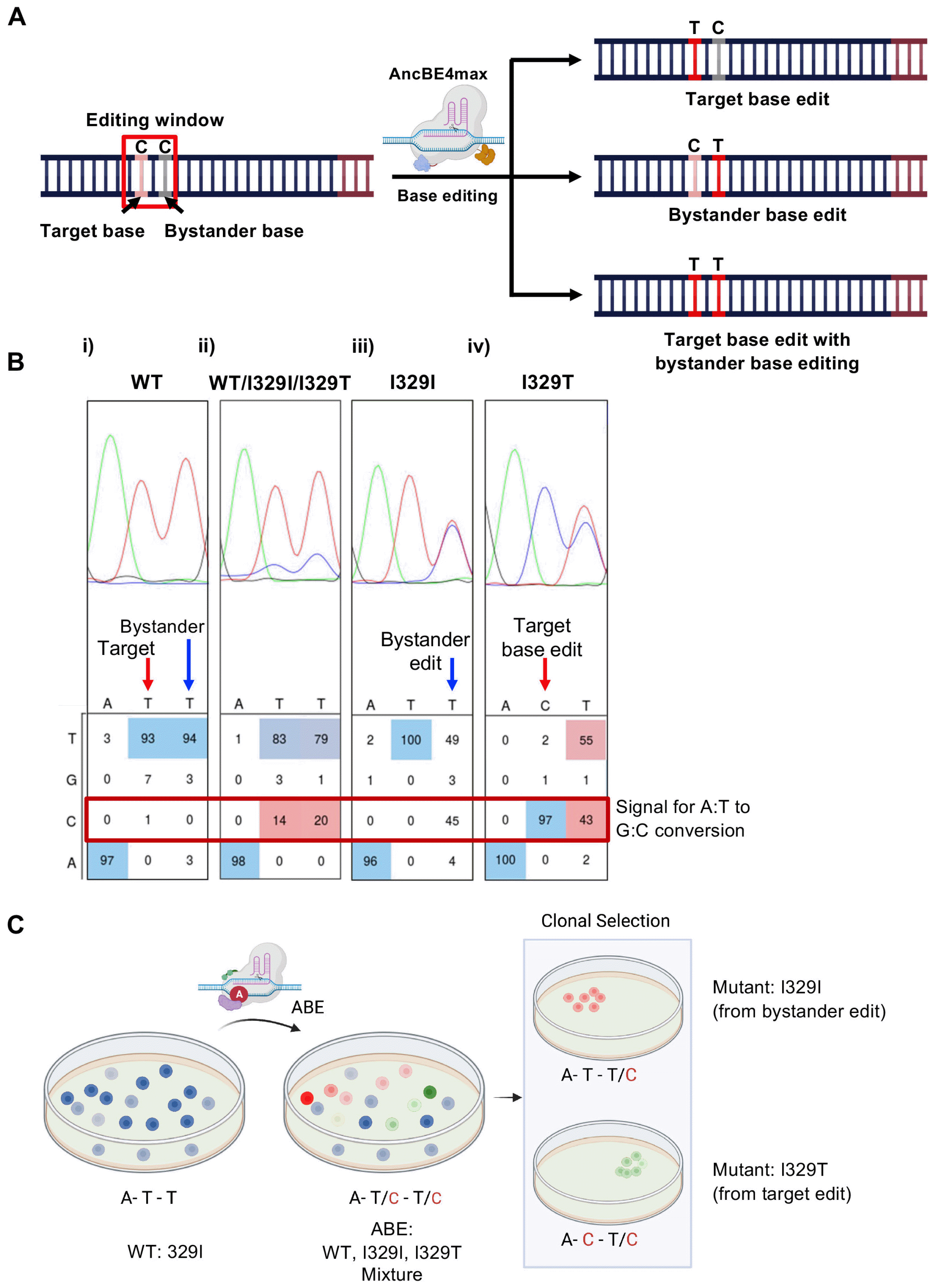 | Fig. 4Bystander base editing of AncBE4max. (A) Graphical scheme for the bystander base in the editing window (colored in red) and possible outcomes. (B) Sequences of GNE encoding 329 isoleucine (329I) in WT hESCs (i), GNE mutants hESCs after ABE application (ii), isolated hESCs with I329I silence mutation due to bystander editing (blue arrow) and GNE mutant hESCs with I329T mutation from target base edit (red arrow) (C) Graphical scheme for GNE mutant hPSCs, WT (blue), I329I mutant (red), and I329T mutant hESCs (green). Created with BioRender.com.
|
Materials and Methods
Single guide RNA (sgRNA) vector synthesis
Equipment
Alcohol Lamp.
Spreader.
42℃ Heat block.
36℃ shaking incubator.
1L erlenmeyer flask.
Polymerase chain reaction (PCR) equipment (Agillent Tcehnologies, SureCycler 8800).
-
Micropipette.
500 ml beaker.
Gel electrophoresis equipment (Takara, AD110).
Agarose gel tray (Takara, AD210).
100 ml Erlenmeyer flask.
Microwave machine.
Autoclave.
1.5 ml E-tube (Axygen, MCT-150-C).
50 ml centrifuge tube (SPL, 50050).
100 mm petri dish (SPL, 10090).
Micropipette tip P10 (Neptune, BT10XLS3).
Micropipette tip P20 (Neptune, BT20).
Micropipette tip P200 (Neptune, BT200).
Micropipette tip P1000 (Neptune, BT1250).
10 ml serological pipette (Corning, 4101).
Latex glove (Ultratex, UF).
Reagents
50x TAE buffer (Biosesang TR2002-100-00).
BsaI (Enzynomics, R0725).
T4 DNA ligase (Solgent, SDL01-R40k).
T4 PNK (Enzynomics, M0055).
10x T4 DNA ligase buffer (Solgent, SDL01-R40k).
Taq DNA polymerase (Solgent, STD95-E500).
10x Taq buffer (Solgent, STD22-B12h).
10 mM dNTP (Solgent, SDN12-B10h).
Ultra pure water (Biosesang, WR4006-100-00).
CIP (BioLabs #M20905).
LB broth stick (LPS, LB-250).
LB agar broth powder (BioSesang LR3004-250-02).
Fragment DNA purification kit (iNtRON, 17290).
Agarose (Young Sciences, Y50004).
Red safe (iNtRON, 21141).
100 bp DNA ladder (iNtRON, 24073).
1,000 bp DNA ladder (iNtRON, 24074).
Ampicillin (BioSesang, AC1043-005-00).
6x DNA buffer (iNtRON, 21162).
Xtra Midi EF Kit (NucleoBond, 740420.50).
8-well Strip Plates (Thermo Fisher Scientific, 15031).
pRG2 vector (addgene, 104174).
DH5α E.coli (Real Biotech, RH617).
pRG2_Rvs primer (5’ gagtcagtgagcgaggaagc 3’).
99% ethyl alcohol (DUKSAN, UN1170).
hPSCs culture, transfection and single cell line establishment
Equipment
Clean bench.
CO2 incubator (37℃, 5% CO2) (Thermo Scientific, 311).
Microscope.
Centrifuge machine.
Hemocytometer.
-
Micropipette.
NEPA21 electroporator (NEPAGENE, NPG-NEPA).
Cell freezing container (NALGENE, 5100-0001).
1.5 ml E-tube (Axygen, MCT-150-C).
60 mm cell culture dish (Corning, 353802).
6 well cell culture dish (FALCON, 353046).
24 well cell culture dish (FALCON, 353047).
Micropipette tip P10 (Neptune, BT10XLS3).
Micropipette tip P20 (Neptune, BT20).
Micropipette tip P200 (Neptune, BT200).
Micropipette tip P1000 (Neptune, BT1250).
10 ml serological pipette (Corning, 4101).
5 ml serological pipette (Corning, 4051).
Latex glove (Ultratex, UF).
Electroporation tube (NEPAGENE, EC-002S).
Reagents
StemFit04 complete media (AJBASIC04CT).
Y-27632 (Sigma, Y0503-5MG).
iMatrix (891-012, ATRIXOME).
Accutase (562527, BD).
Opti-MEM (11058021, gibco).
AccuPrep genomic DNA extraction kit (BIONEER, K-3032).
Dulbecco’s phosphate buffered saline (DPBS) (Gibco, 14190-250).
Taq DNA polymerase (STD95-E500).
10x Taq buffer (STD22-B127).
10 mM dNTP (SDN12-B10h).
ABE8e vector (addgene, 138489).
NG-ABE8e vector (addgene, 124163).
AncBE4max vector (addgene, 112094).
YE1-BE4max-NG vector (addgene, 138159).
siRNA targeting UNG (BIONEER, 7374-2).
Procedure
Designing and cloning sgRNA
Material Preparation
-
Preparation of LB broth with ampicillin (100 μg/ml).
- Dissolve ampicillin powder to 100 mg/ml with UPW.
- Add two LB broth powder stick and 500 ml of distilled water (DW) to 1 L Erlenmeyer flask.
- Wrap top of 1 L Erlenmeyer flask with aluminum foil.
- Autoclave LB broth and cool down to room temperature.
- Add 500 μl of ampicillin (100 mg/ml) in front of flamed alcohol lamp (Note. Flame micropipette tip before use).
-
Preparation of LB agar broth plate with ampicillin (100 μg/ml).
- Add LB agar broth powder 20 g and 500 ml of DW in 1 L Erlenmeyer flask.
- Wrap top of 1 L Erlenmeyer flask with aluminum foil.
- Autoclave LB agar broth and cool down to 40∼50℃ then add 500 μl of ampicillin (100 mg/ml) in front of flamed alcohol lamp (Note. Flame micropipette tip before use).
- Add 15 ml of LB agar broth to 100 mm petri dish in front of flamed alcohol lamp (Note. Flame serological pipette before use) and set in room temperature for overnight.
sgRNA cloning
Editing tool for C:G to T:A or A:T to G:C: Base editor is selected as described in Figure 2 among AncBE4max (editing window: 4∼8 nt), NG-AncBE4max (editing window: 4∼8 nt), ABE8e (editing window: 4∼8 nt), and NG-ABE8e (editing window: 4∼7 nt).
Order DNA oligo for sgRNA cloning: Add CACC sequence to 5’ of forward oligo and AAAC sequence to 5’ of reverse oligo as Table 1 in order to match the sticky end of pRG2 vector cleaved by BsaI.
-
Preparation of Insert.
- Dilute each oligo to 10 pmol/μl.
- Add 1 μl of each oligo, 1 μl of 10x T4 ligation buffer, 1 μl of T4 PNK, 6 μl of UPW to 8-well strip plate.
- React as Table 2 follow with PCR machine (melt and re-annealing).
- Dilute reacted solution to UPW with 1/50 ratio in e-tube.
-
Digestion of pRG2 vector with enzymes.
-
Preparation of Enzyme-digested pRG2 vector.
- Dilute 50x TAE buffer to 1x concentration with UPW.
- Mix 1x TAE buffer and agarose for 1% (weight/volume) agarose gel in 100 ml Erlenmeyer flask.
- Melt agarose with microwave, then add red safe and mix gently by shaking flask.
- Set agarose gel in gel tray.
- Mix solution from 1.2.d with 6X DNA loading buffer. Load the solution into a well of 1% agarose gel, and 1,000 bp DNA ladder into other well of the gel.
- Set the gel into electrophoresis equipment and run the equipment.
- Cut the gel at 2,500 bp position, checking a clear band being loaded within the cut gel fragments. Also, check the unclear band at 700 bp position within uncut part of the gel.
- Extract DNA from the cut gel fragment with DNA purification kit.
Add 0.7 μl of solution from 1.2.c, 30 ng of purified DNA from 1.2.e, 1 μl of 10×ligation buffer, 1 μl of T4 DNA ligase, and UPW up to 10 μl to 8-well strip plate, react at 16℃ for overnight.
Add 5 μl of solution from 1.2.f to 50 μl of DH5α E.coli, then, heat-shock at 42℃ for 1 minute (min). (Note. Keep DH5α in ice).
Place DH5α from 1.2.g in ice for 10 min.
Spread DH5α from 1.2.h to LB agar plate with ampicillin by spreader. (Note. Keep spreader in 99% EtOH, flame before spread DH5α).
Incubate the LB agar plate from 1.2.i at 37℃ for overnight.
-
Colony PCR.
- Inoculate colony formed on LB agar plate from 1.2.m to 1 ml LB broth with ampicillin for 4 hr in 37℃.
- Add 1 μl of DH5α, 2.5 μl of 10×Taq buffer, 0.25 μl of Taq DNA polymerase, 0.5 μl of dNTP, 2 μl of forward oligomer from 1.2.b, 2 μl of pRG2_Rvs primer, and 16.75 μl of UPW to 8-well strip plate. PCR as Table 3.
Mix PCR product with 6×DNA buffer and run with 100 bp DNA ladder in 2 % agarose gel by gel electrophoresis equipment. If DNA band with 250 bp size is observed, sgRNA cloning can be considered as completed.
Add DH5α from 1.2.k to 500 ml of LB broth with ampicillin, culture in 37℃ with shaking incubator for overnight.
Prep sgRNA vector from cultured DH5α with Xtra Midi EF Kit. Sequence prepped plasmid DNA with pRG2_Rvs primer for confirmation of cloning.
Table 1
| F/R | 5’ add | 20nt spacer sequence without PAM (5’ to 3’) | |
|---|---|---|---|
| Gene of interest | F | CACC | GN1N2…Ni (i=19 or 20) |
| R | AAAC | NiNi-1…N1C |
The N represents nucleotide A, T, G, or C. The red Ni represents reverse complementary sequence of the Ni. Note that a spacer sequence must start with G, because transcription driven by U6 promoter starts with G. In this case, the produced sgRNA is called GX19, which has 20 nucleotides of spacer sequences with 5’ G matched with target site. If a spacer sequence does not start with G, additional G nucleotide must be added upstream of spacer sequence for appropriate transcription. In this case, the produced sgRNA is called GX20 or gX20, which has 20 nucleotides of spacer sequences with additional 5’ G matched or unmatched with target site.
![]()
hPSCs culture, transfection and single cell line establishment
hPSCs culture and transfer
Among various hPSCs culture media and coating materials, we adopted StemFit 04 complete media and iMatrix for clinical grade culture condition.
When cells grew up to 70∼80% of confluency, suction culture media and add 1 ml of Accutase for 60 mm cell culture dish, 500 μl for 6 well cell culture dish, and 150 μl for 24 well cell culture dish. Incubate at 37℃ for 10 min in CO2 incubator.
Transfer detached cells to e-tube, centrifuge with 1,000 rpm for 1 min at room temperature.
Suction supernatant, wash with DPBS for two times.
Resuspend cells with 200 μl of Y27632 10 μM culture media.
Seed 5∼20 μl of cells to well plate with cell transfer culture media.
Wash out cell transfer culture media with DPBS and add cell culture media after 1 day from transfer.
Change media every other day.
hPSCs transfection
Transfection requires 1×106 cells.
Detach and wash cells followed by process 2.2.b∼2.2.d.
Resuspend cells with opti-MEM and count with hemocytometer.
Dilute cells to 1×106 cell/100 μl.
Add 3.5 μg of base editor vector, 1.5 μg of sgRNA vector to 100 μl of cell (1×106 cell/100 μl). (Note. For CBE application, add 2 μg of siRNA targeting UNG to increase editing efficiency and product purity).
After pipetting, transfer solution from 2.3.e to electroporation tube.
Electroporate cell with NEPA 21 with condition described in Table 4. (Instead of NEP21, other electroporation system, such as Amaxa 4D Nucleofector system (Lonza) was also used as described (31)).
Transfer electroporated cells to e-tube with 200 μl of Y27632 10 μM culture media, using pipette contained within electroporation tube set.
Seed cells to well plate with cell transfer culture media.
Single cell picking
After 4 days from transfection, detach and wash cells followed by the previous step from 2.2b to 2.2e.
Count cells with hemocytometer and dilute 1 cell/1 μl.
Seed 10 μl of diluted cells from 2.4.b to 60 mm culture dish with cell transfer culture media. Change media every other day.
After colony formation (usually takes 1∼2 week), mechanically detach each colony with 1 ml micropipette tip, and transfer to e-tube with 50 μl of cell transfer culture media.
After breaking colony with pipetting, transfer cells to 24 well plate filled with cell transfer culture media. Culture up to 10∼30% confluency.
Colony genotyping
Transfer cells from 2.4.e followed by process 2.2.b∼2.2.f.
Extract genomic DNA from left cells from 2.5.a with AccuPrep genomic DNA extraction kit.
Design primer for target gene amplification with primer3web (https://primer3.ut.ee/).
-
Add 1 μl of gDNA, 2.5 μl of 10×Taq buffer, 0.25 μl of Taq DNA polymerase, 0.5 μl of dNTP, 2 μl of forward primer, 2 μl of reverse primer, and 16.75 μl of UPW to 8-well strip plate. PCR as follow.
Analyze amplified target gene with Sanger sequencing.
 XML Download
XML Download