

 PDF
PDF Citation
Citation Print
Print


Introduction
The heart contains a heterogenous pool of cardiac progenitor cells (CPCs). These different populations of CPCs, such as the c-kit+ (1) and Sca1+ cells (2) have previously been demonstrated to exert a role in cardiac growth, development, and repair. c-kit+ cells functioning as cardiovas-cular precursors have been detected in adult human hearts undergoing hypertrophy, post-myocardial infarction and post-ischemia/reperfusion injury, albeit the number of the c-kit+ cells may differ depending on the cardiac condition (3) and origins in the different heart chambers (4). Infusion of CPCs into the injured myocardium has been demonstrated to result in cardiac restoration and improved function (5). Yet lineage tracing studies to track the differenti-ation of c-kit+ cells into cardiomyocytes have been contradic-ting, depending on the experimental system employed (6). Irrespective of the contradictory findings, studies conducted in general have suggested that c-kit+ CPCs can be found in the hearts of different animals ranging from the mice, pig, and sheep to the human heart at different developing stages (7). Similarly, regardless of whether the beneficial effects of CPCs infusion may purely be due to invoking certain indirect paracrine mechanisms (8), c-kit+ cells exert a certain level of role in functional cardiac regeneration and repair (9).
Recent studies conducted have alluded to the role of mitochondria, particularly the influence of mitochondrial dynamics in the differentiation process of induced pluripo-tent stem cells (iPSCs) (10) and the embryonic stem cells (ESCs) (11) whereby mitochondrial fusion, in general, has been demonstrated to be crucial for stem cell differen-tiation. The elongation of the mitochondria coincides with the development of cristae, reflecting increased mitochondrial activity in the differentiated cell (12). In the quiescent adult cell, the mitochondria exist in a dynamic equilibrium between an elongated (via mitochondrial fusion) versus a fragmented (via mitochondrial fission) phenotype (13, 14). The fusion of individual mitochondrions is governed by pro-fusion proteins such as Mitofusins 1 or 2 (Mfn1 or 2) positioned on the outer mitochondrial membrane (OMM) (15) and Optic Atrophy 1 (OPA1) on the inner mitochondrial membrane (IMM) (16). The fragmentation of the mitochondrion depends on the Dynamin-related Protein 1 (Drp1), which translocates from the cytosol to the mito-chondrion upon activation by calcineurin – a calcium and calmodulin dependent serine/threonine protein phosphatase (17). The change in mitochondrial morphology is affected by the physiological environment in which different cues such as intracellular calcium or ROS levels may tilt the mitochondrial morphology towards a fused or fragmented state. Similarly, the removal of dysfunctional mitochondria via autophagy/mitophagy or a shift in cellular metabolism affects the fusion and fission process of the mitochondria which ultimately dictate cellular fate.
In this study, we investigated the changes in mitochondrial morphology in the dexamethasone-mediated differentiation process of c-kit+ CPCs. We discovered that mitochondrial fragmentation precedes the differentiation of cardiac c-kit+ CPCs and inhibition of mitochondrial fragmentation abrogates the differentiation status of c-kit+ CPCs. In this regard, our observations highlight a potential role of mitochondrial morphology in the differentiation of cardiac c-kit+ CPCs, whereby pharmacological modulation of mitochondrial morphology may dictate cardiac progenitor cell fate in the heart.
Materials and Methods
CPC isolation and culture
All experimental procedures were performed in accordance with institutional guidelines and approved by the Ani-mal Experimentation Ethics Committee of the Chinese Uni-versity of Hong Kong. c-kit+ CPCs were isolated from 2-month-old male wild-type FVB mice. All c-kit+ adult CPCs were isolated and cultured as previously described (18). To activate differentiation, CPCs were incubated in α-mini-mal essential medium containing 10 nM dexamethasone for up to 7 days (19). To inhibit Drp1-mediated mitochondrial fragmentation, either 10 μM or 50 μM mdivi-1 (Sigma) was administered once at Day 0 and again at Day 2 of differentiation (20). To inhibit calcineurin, either 1 μM or 5 μM ciclosporin-A (CsA) (Sigma) was administered once at Day 0 and again at Day 2 of differentiation (17).
Immunofluorescence microscopy
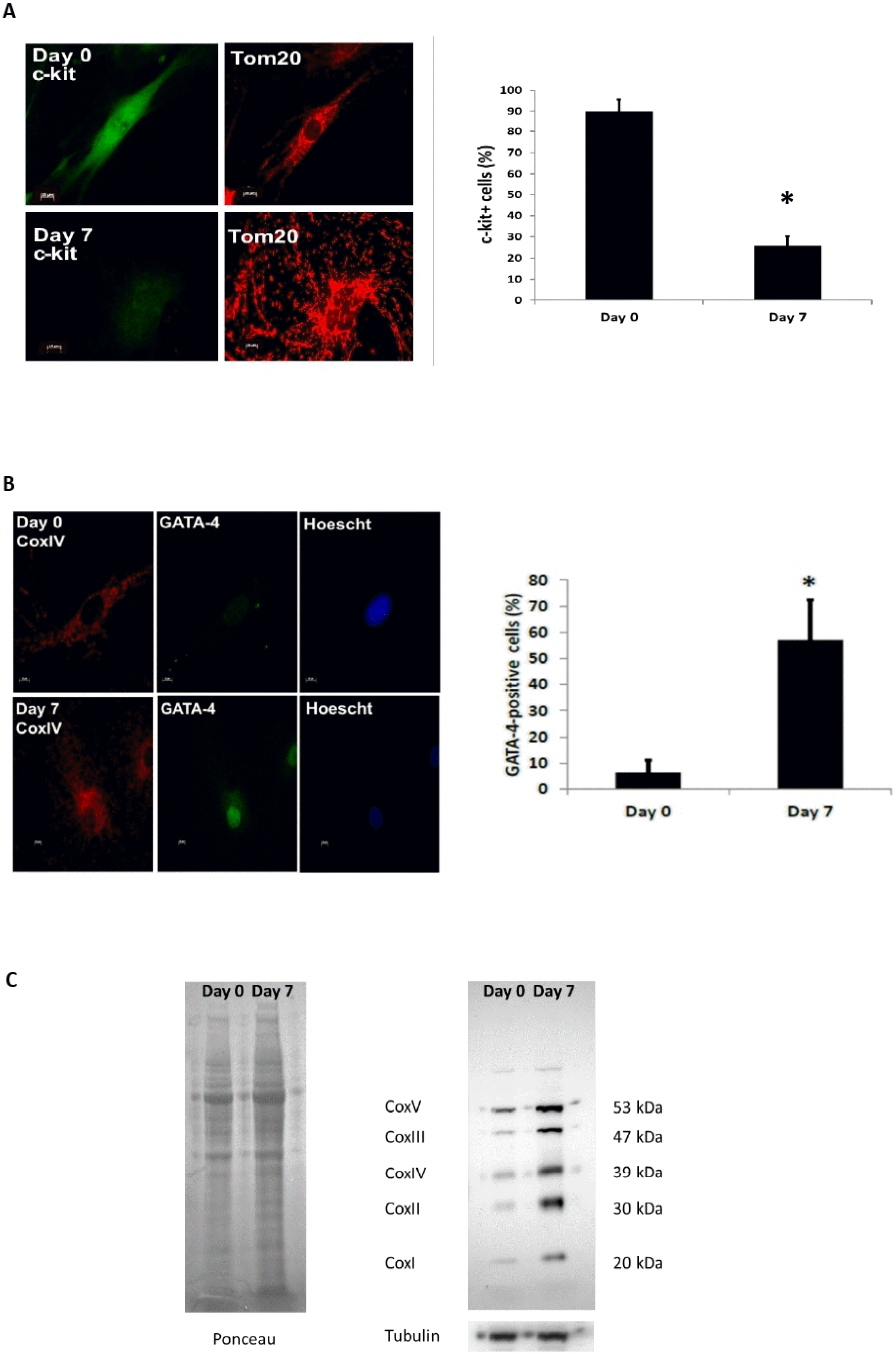
Cells were fixed, permeabilized, and blocked as previously described (19). CPCs were stained with antibodies against Tom20 (Santa Cruz Biotechnology), COXIV (Invi-trogen), and GATA-4 (Santa Cruz Biotechnology). Cells were incubated with Alexa Fluor 488 or 594 secondary antibodies (Life Technologies) followed by Hoechst 33342 (10 μg/ml, Life Technologies) to stain nuclei. Cells were imaged using a Carl Zeiss Axio Observer Z1, and Z-stacks were acquired using a high resolution AxioCam MRm digital camera, a 63× oil immersion objective, and Zeiss Axio-Vision 4.8 software (Carl Zeiss). The number of mitochondria within each image and the average size of each mitochondrion (total mitochondrial length over the total number of mitochondria) was quantified using ImageJ. A minimum of 80 cells per group were counted for each condition.
Western blot
Samples were prepared in lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, and protease inhibitor mixture (Roche Applied Science) and run on Invitrogen NuPAGE Bis-Tris gels. The membranes were probed with the following antibodies: MitoProfile Total OXPHOS Rodent WB antibody mixture (MitoSciences), Opa1 (Abcam), Mfn1 (Santa Cruz Biotechnology), Mfn2 (Sigma), Drp1 (BD Biosciences), Fis1 (Sigma), Tubulin (Sigma), Drp1 (phospho S637) (Abcam). Membranes were imaged using a ChemiDoc XRS+ System (Bio-Rad).
Calcineurin activity assay
The calcineurin phosphatase activity was measured by the dephosphorylation rate of a synthetic phosphopeptide substrate with a calcineurin assay kit (Enzo Life Sciences, Plymouth Meeting, Pa, USA) following the manufacturer’s instructions. The released free phosphate was then detected colorimetrically with the green reagent on a plate reader at 620 nm.
Results
Dexamethasone mediates the differentiation of c-kit+ CPCs
Incubation of c-kit+ CPCs with dexamethasone for a period of 7 days activates the differentiation process as evidenced by the loss of c-kit staining (Fig. 1A) and appearance of the cardiac myocyte lineage marker GATA-4 at Day 7 post-dexamethasone treatment (Fig. 1B). The 7 days of dexamethasone treatment also increases the expression of the mitochondrial respiratory subunits (Fig. 1C).
Dexamethasone-mediated c-kit+ CPCs differentiation is aligned alongside mitochondrial fragmentation
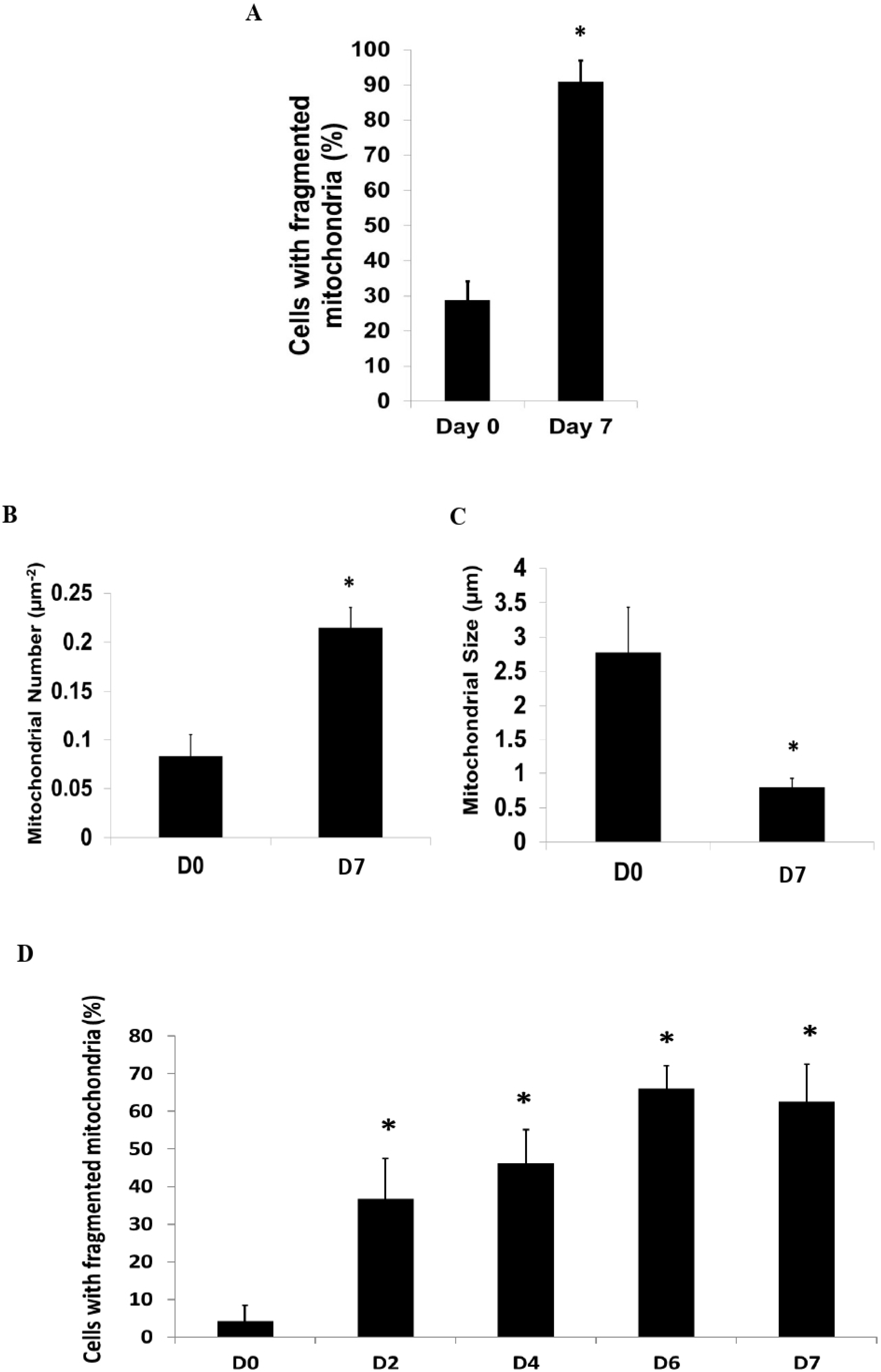
In addition to the loss of c-kit and the appearance of GATA-4 following differentiation, we observed a signifi-cant increase in the proportion of c-kit+ CPCs with fragmented mitochondria (Fig. 2A). In addition, the number of individual mitochondrions was significantly increased (Fig. 2B), whilst the average mitochondrial size was significantly reduced (Fig. 2C) following dexamethasone-mediated differentiation for 7 days. The fragmentation of the mito-c-hondria started as early as Day 2 following dexametha-sone treatment (Fig. 2D).
Calcineurin-Drp1 mediates mitochondrial fragmentation during differentiation of c-kit+ CPCs
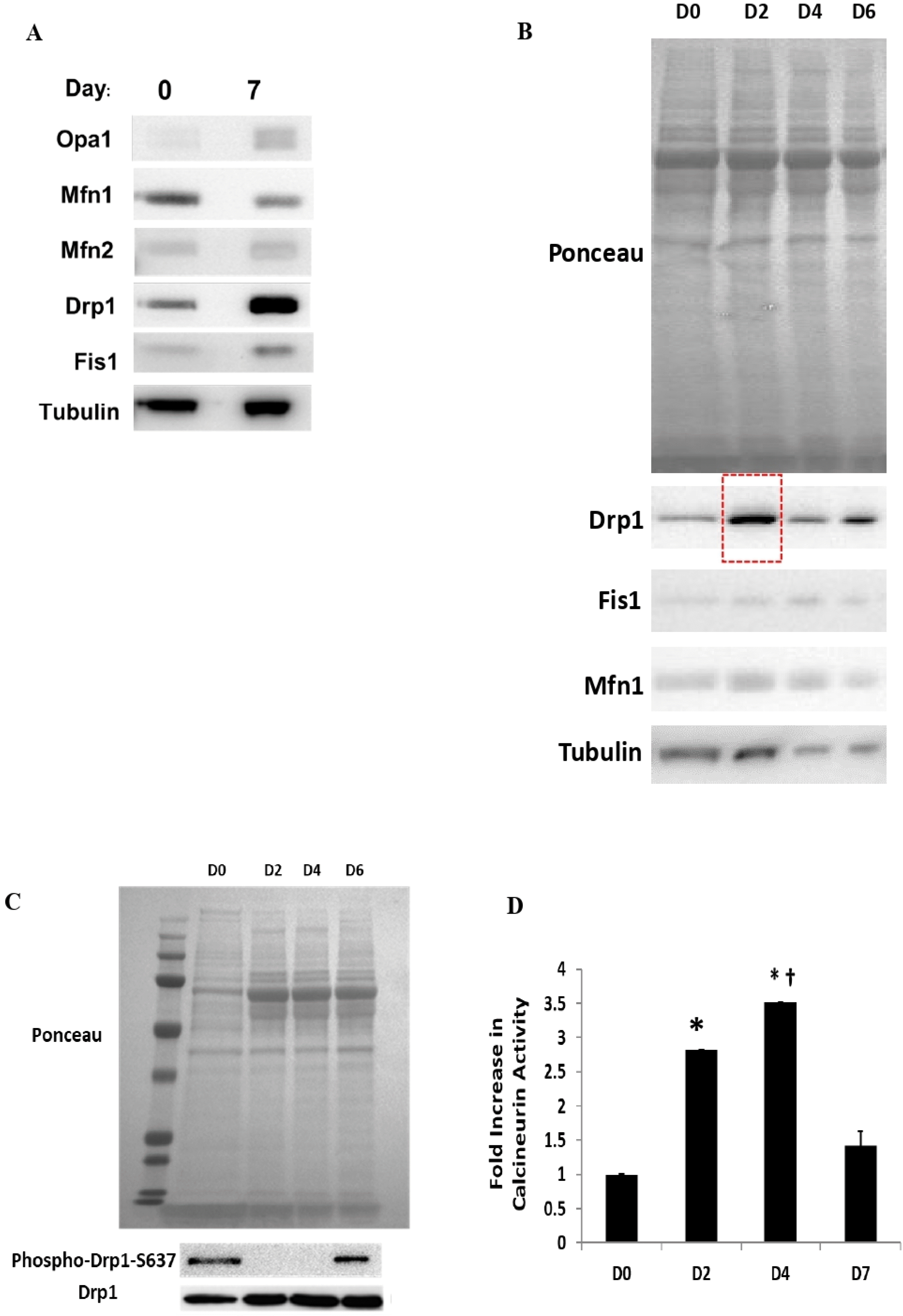
We next assessed the mitochondrial-shaping protein responsible for mediating the shift in mitochondrial morphology to a fragmented state during the differentiation process. We observed that the level of Drp1 was signifi-cantly upregulated at Day 7 post-differentiation (Fig. 3A). This increase in Drp1 protein expression started as early as Day 2 following dexamethasone treatment (Fig. 3B) whilst de-phosphorylation of Drp1 at the site of Ser637 (S637) occurred on both Days 2 and 4 post-dexamethasone treatment (Fig. 3C). As Drp1 activation is known to be mediated upstream by calcineurin, we employed the use of a calcineurin activity assay kit to determine calcineurin activity (Fig. 3D). We found that the calcineurin activity was increased significantly at Day 2 post-dexamethasone treat-ment. The calcineurin activity increased further at Day 4, albeit not significantly compared to Day 2. At Day 7, the calcineurin activity reduced to a level which was almost similar to Day 0.
Inhibiting Drp1 abrogates mitochondrial fragmentation and impairs CPC differentiation
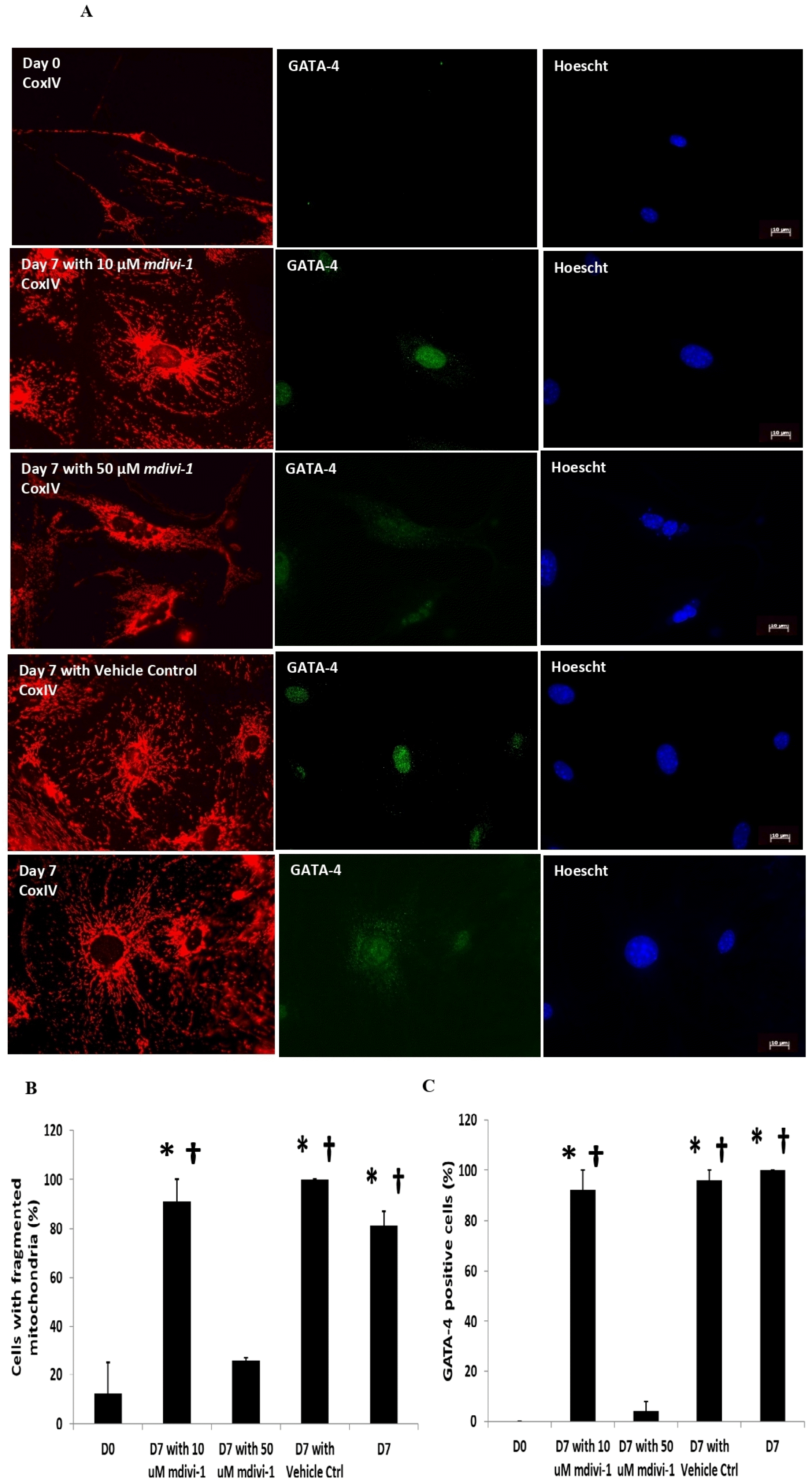
As the fragmentation of the mitochondria correlates with an increase in total Drp1 levels, we investigated whether inhibiting Drp1 affects the differentiation of c-kit+ CPCs. We subjected the cells to 2 different doses of mdivi-1 −10 μM or 50 uM; once at Day 0 and again at Day 2. We found that the presence of 50 μM mdivi-1 significantly reduced the proportion of c-kit+ cells with predominantly fragmented mitochondria following 7 days of dexamethasone treatment (Fig. 4A and 4B). The proportion of GATA-4-positive c-kit+ CPCs at Day 7 were also significantly reduced following the 50 μM mdivi-1 treatment (Fig. 4A and 4C). 10 μM mdivi-1, however, failed to inhibit mitochondrial fragmentation and reduce the number of GATA-4 positive cells.
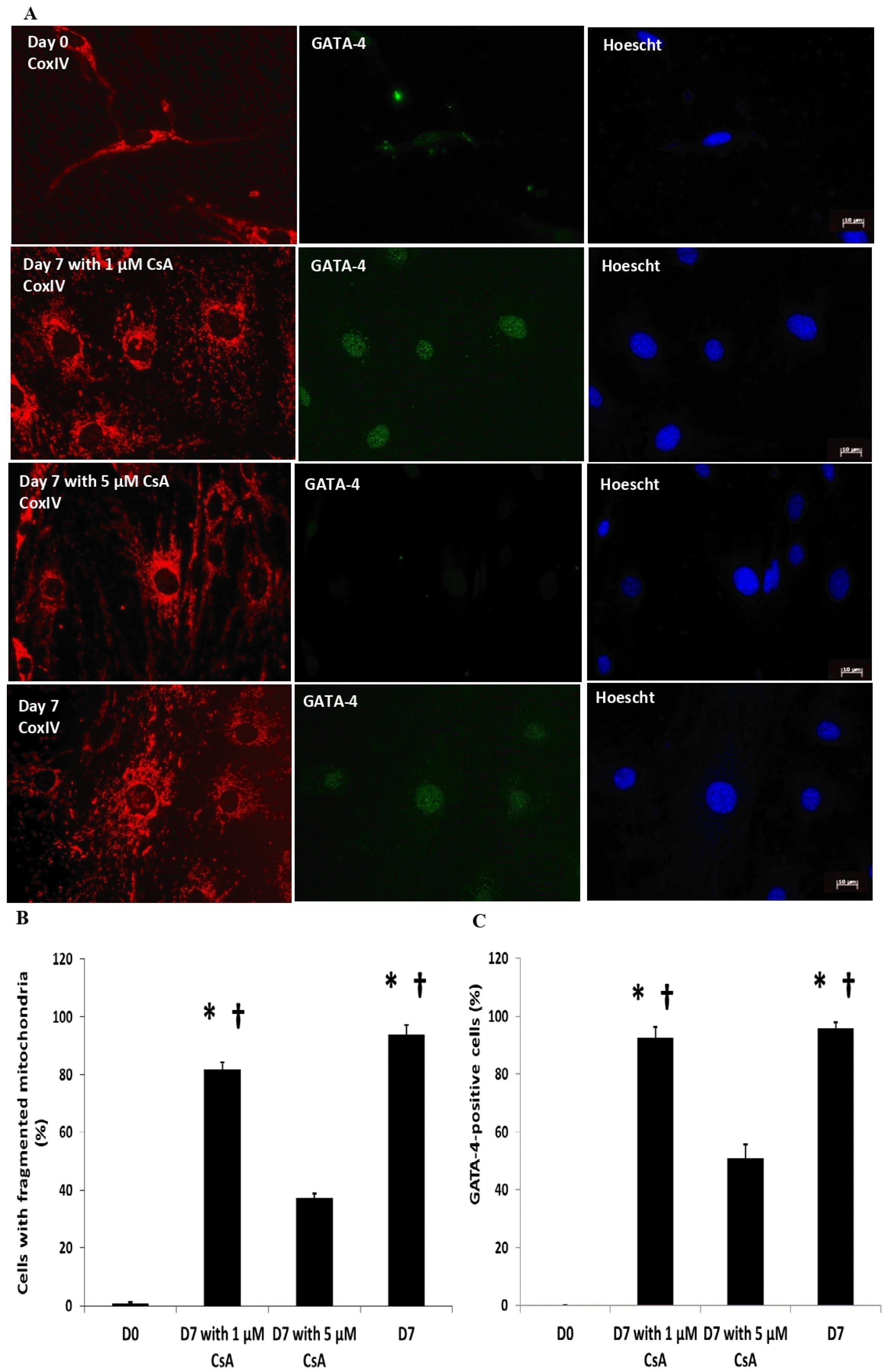
Inhibiting calcineurin abrogates mitochondrial fragmentation and impairs CPC differentiation
Elevated calcium has been known to activate calcineurin leading to de-phosphorylation of Drp1 and subsequent translocation from the cytosol to the mitochondria for execution of mitochondrial fragmentation. We sought to investigate whether modulating calcineurin upstream of Drp1 via CsA at 2 different doses (1 μM or 5 μM) impedes the differentiation of c-kit+ CPCs. Inhibition of calcineu-rin using 5 μM CsA but not 1 μM CsA, significantly reduced the proportion of c-kit+ CPCs with fragmented mitochondria (Fig. 5A and 5B). Consequently, the propo-rtion of GATA-4-positive c-kit+ CPCs was also signifi-cantly reduced by 5 μM CsA as opposed to using 1 μM CsA (Fig. 5A and 5C).
Discussion
The study conducted here demonstrated that dexame-thasone-induced differentiation of c-kit+ CPCs is aligned with Drp1-mediated mitochondrial fragmentation. Inhibition of Drp1 using the small molecule inhibitor of Drp1 – mdivi-1 or upstream calcineurin via CsA impairs the differentiation process. Thus, our findings allude to the impor-tance of mitochondrial fragmentation in the dexamethasone-mediated differentiation process of cardiac c-kit+ CPCs.
Our study utilised dexamethasone for a total of 7 days to induce differentiation according to previous literature (19). Although not included in this study, we have also demonstrated that the dexamethasone-mediated differentiation process of c-kit+ CPCs led to upregulation of other cardiac lineage markers such as GATA-6, MEF2C, as well as enhanced mitochondrial oxygen consumption rates, consistent with our previous study (19). In the current study, dexamethasone-induced differentiation of c-kit+ CPCs promoted a reduction in levels of c-kit expression, an increase in GATA-4 expression aligned with Drp1-mediated fragmentation of the mitochondria. Our results are consistent with the findings of other studies whereby the use of pharmacological nitric oxide (NO) donors to increase mitochondrial numbers promotes maturation of fast-dividing mesoangioblasts into cardiomyocytes whilst decreasing mitochondrial content using respiratory chain inhibitors and chloramphenicol perturbs cardiomyocyte differentiation in slow-dividing populations (21). Rounding of the mitochondria was also observed in the aggregation stage of P19 cells differentiation into cardiomyocytes (22). Knock-down of Drp1 negatively influences 30-days neurogenesis of ESCs, coincident with a delayed reduction of Oct4 and Nanog during the mid-differentiation process (23). None-theless, the consensus derived from studies involving iPSCs and ESCs is that mitochondria in stem cells are immature, perinuclear-localized, fragmented, with fewer cristae (24). Upon differentiation to terminal cell types, the mitochondria become enlarged, elongated, and tubular concomitant with an increase in mitochondrial content (24). A low mito-chondrial mass has been associated with the undifferen-tiated and rapidly dividing state of ESCs and iPSCs (25, 26). Lowering MFN2 and OPA1 levels by gene trapping impaired differentiation of ESCs into beating cardiom-yocytes (26). Nuclear reprogramming by the stemness transcription factors - OCT3/4, SOX2, KLF4, and c-MYC restructured the mature tubular and cristae-rich somatic mitochondria in mouse embryonic fibroblasts (MEF) into immature spherical and cristae-poor structures in iPSC (27). The presence of these stemness transcription factors induced an increase in phosphorylation of ERK1/2 and DRP1-S579, thus driving Drp1-mediated mitochondrial fragmentation (28). Administration of 50 μM mdivi-1 on day 3 before the appearance of iPSC colonies in MEFs transduced with individual lentiviruses containing OCT4, SOX2, and KLF4 was sufficient to reduce somatic cell reprogramming by >95% 14 days post-viral transduction (25). Mfn2 has also been demonstrated to regulate mammalian postnatal male germ cell development by modulating both mitochondrial and ER functions – a process which is distinct from Mfn1 which only regulates mitochondrial fusion during neonatal pro-spermatogonia (29). A comparison between these different studies alluded to the fact that the discrepancy in the observation of mitochondrial fusion-fission during differentiation may be due to different confounding factors such as the source of progenitor cells, different agents used for induction of differentiation, contrasting differentiation markers, varying di-fferentiation durations, and desired terminal cell types.
Although the level of intracellular calcium (Ca2+) was not investigated in this study, electrical stimulation in human c-kit+ CPCs has been found to activate a Ca2+-dependent signaling pathway in which the increased intracellular Ca2+ activates the transcription of cardiospecific genes and proper alignment of the cells in a tissue-like structure, albeit a direct investigation into the lineage fate was not examined (30). An increase in free intracellular Ca2+ in canine c-kit+ CPCs revealed a prominent inwardly rectifying current identified as the intermediate conductance Ca2+‐ activated K+ current (KCa3.1) that activates progenitor cell proliferation, although this was not extrapolated to the differentiation of c-kit+ CPCs (31). During cardiomyogenesis, activation of the TGFβ receptor regulates TAK, which then acts on the transcription factors ATF2 and CREB through the Ca2+ responsive MAPK pathway (32). In addition, adipogenic differentiation mediated by a combination of dexamethasone, indomethacin, and insulin in human umbilical cord blood-derived MSCs (hUCB-MSCs) has been found to be enhanced by Ca2+ via negative regulation of the Wnt5a/β-catenin signaling pathway (33).
Downstream of the increase in intracellular calcium, calcineurin has been established to be a Drp1-activator (34). Calcineurin activated in a Ca2+-calmodulin dependent fashion dephosphorylates Drp1 at Ser637, thus mediating the translocation of Drp1 from the cytosol to the mitochondria for activation of the scission process (17). We found that inhibition of calcineurin via CsA abrogates mitochondrial fragmentation and impairs the differentiation process of c-kit+ cells. Mirroring our results, the calcineu-rin-NFAT signaling has also been demonstrated to converge on the Erk1/2 pathway to regulate Src expression, which is crucial for lineage specification in response to varying differentiation stimuli such as LIF-withdrawal or retinoic acid addition in mouse ESCs (mESC) (35). This finding was further supported by the finding that the alternative splicing regulator MBNL1 promotes generation of the atypical calcineurin Aβ variant CnAβ1, which is localized to the Golgi apparatus and regulates the intracellular localization and activation of the mTORC2 complex, thus driving differentiation in mESCs (36). Calcineurin signaling has also been associated with transforming growth factor (TGF)-β1, which drives the differentiation of neural crest stem cells (NCSCs) into smooth muscle (37).
There are certain limitations in our study, one of which is that our differentiation process was not extended beyond 7 days. Our current protocol of dexamethasone treatment for 7 days is sufficient to induce differentiation of c-kit+ CPCs as evidenced by the expression of different cardiac lineage markers (19), in line with the findings of other previous studies (38). Yet, the differentiation process in certain studies involving iPSC-CMs and ESC-CMs had an extended duration until approximately 12∼15 weeks, whereby the structural, electrophysiological, and mechanical properties of these late stage cells exhibit a higher similarity to that of an adult cardiomyocyte (39), although it should be noted that the maturation of the mitochondria remains inadequate and suboptimal compared to fetal development despite the increase in relative mitochondrial abundance or mass following extended culture (39).
Apart from dexamethasone, the differentiation process of c-kit+ CPCs can also be initiated by different cues such as (i) TGF-β, which has been shown to result in differentiation to myocytes, (ii) dexamethasone and VEGF for differentiation into endothelial cells and (iii) dexame-thasone together with growth factor PDGF BB or all-trans retinoic acid for differentiation into smooth muscle cells. Furthermore, the detected transcription factors for the formation of either myocytes, endothelial cells, or smooth muscle cells may also explain the varying levels of differentiation observed in different studies (40). In addition, studying the electrical activity of the differentiated cells will reveal more about the physiological attributes of the cells (30). Calcineurin inhibition using alternative CsA analog such as FK506 (Tacrolimus) or CN585 should be performed in future to confirm the association of calcineurin-mediated de-phosphorylation of Drp1 to induce mitochondrial fragmentation as there are calcineurin-independent effects of CsA which includes the cyclophilins (41, 42), cardiac actin expression (43), mitochondrial Ca2+ homeostasis (44), ROS elevation (45), ER stress (46) and even TGF-beta production (47). Pharmacologically inhibiting Drp1-mediated mitochondrial fragmentation by other alternatives such as P110 or genetic silencing of Drp1 will also help in further validating the results as mdivi-1 may have other Drp1-independent off-target effects such as inhibition of mitochondrial respiratory Com-plex I (48). Comparing the c-kit+ CPCs isolated from other strains of mice such as the heterozygous Drp1-KO mice will also allow further investigation into the relevance of Drp1-mediated mitochondrial fragmentation in the differentiation process of c-kit+ CPCs. Similarly, whether other pro-fission regulators such as Mff or MiD49/51 exert a role in the differentiation of c-kit+ CPCs remains to be determined.
Taken together, the findings from this study highlight the pertinence of mitochondrial morphology in differentiation status of progenitor cells and potentiates pharmacological manipulation of the mitochondria in controlling progenitor cells fate. This is particularly important in the wake of translating progenitor cell research in the fields of regenerative and cardiovascular medicine whereby an optimal spatial positioning and proper temporal differentiation of progenitor cell is crucial.
 XML Download
XML Download