

 PDF
PDF Citation
Citation Print
Print



COMMON MOLECULAR METHODS
While translocation driver events are very rare in epithelial malignancies, they occur in up to 25% of sarcomas. As a result, molecular techniques for identifying recurrent translocations currently play an important diagnostic role in bone and soft tissue pathology.
Fluorescence in-situ hybridization (FISH)
FISH uses a fluorescently labeled probe targeted towards a specific genetic sequence. The fluorescent probe can then be assessed in situ using a fluorescence microscope. Dual-color dual-fusion probes are designed towards a particular gene fusion target, while dual-color break-apart probes have greater utility for gene fusions in which the partner may not be known (e.g. EWSR1). Large gene amplifications and deletions may also be detected by comparing the ratio of lost target signals to a control signal (e.g. MDM2).
Reverse-transcriptase polymerase chain reaction (RT-PCR)
PCR-based tests utilize PCR amplification of certain primer sequences that can be built to accommodate a set of gene rearrangements. This is particularly useful for larger panels that use known genetic breakpoints, but this method generally lacks the ability to detect new fusion partners.
Next-generation sequencing (NGS)
NGS has become standard for detection of both prognostic and therapeutic mutations. In sarcoma, new RNA-based methods, including hybrid-capture and anchored multiplex PCR, offer better detection for gene rearrangements. These methods have the added benefit of detecting new gene partners, which is particularly useful with promiscuous genes like EWSR1. The development of various commercial platforms for NGS fusion testing has significantly increased the availability of this test to pathologists. Additionally, tertiary institutions with large sample volume in bone and soft tissue pathology may opt to bring these methods in-house to improve turnaround times.
Methylation testing
Initially developed for usage in the diagnosis of brain tumors, methylation testing has expanded to begin to include soft tissue and bone tumors. Methylation tests use an array chip to detect the methylation patterns of thousands of CpG islands to produce a “signature” for a particular tumor. Through statistical methods, these signatures can be clustered into groups of similar tumors, providing a tentative “cell of origin” diagnosis (Fig. 1). While still in its infancy, methylation has great potential for future diagnostics in soft tissue and bone.
TESTING GUIDELINES FOR SPECIFIC TUMOR CLASSES
The constantly growing number of translocation-driven soft tissue and bone tumors in the literature necessitates judicious use of diagnostic genetic testing. By employing a morphology-based approach, pathologists can triage mesenchymal neoplasms for both diagnostic and therapeutic testing. The following review takes a “line of differentiation” approach to decide on appropriate testing strategies.
Adipocytic
• MDM2 amplification is the key differentiating factor between lipoma and well-differentiated/dedifferentiated liposarcoma (Fig. 2). MDM2 amplification may also rarely be seen in other high-grade sarcomas; as such, caution is warranted when interpreting this finding without the presence of a well-differentiated liposarcoma component.
• RB1 deletion (tested by loss of RB1 on immunostaining or NGS) is pathognomonic for spindle cell/pleomorphic lipoma and may be seen in about 50% of atypical spindle cell/pleomorphic lipomatous tumor.
• Myxoid liposarcoma is driven by translocations involving DDIT3, most commonly with FUS. These tumors show a distinct myxoid background, chicken-wire capillary architecture, and univacuolated lipoblasts. High-grade variants show round cell features.
Fibroblastic/myofibroblastic
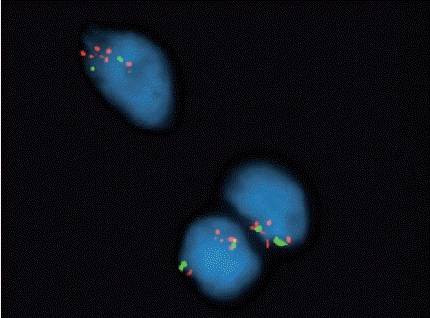
• Nodular fasciitis and similar disorganized, myxocollagenous, variably cellular fibroblastic/myofibroblastic neoplasms with or without reactive bone formation (including myositis ossificans, fibro-osseous pseudotumor of the digit, aneurysmal bone cyst, and a subset of fibroma of tendon sheath), form a growing spectrum of tumors that are defined by USP6 gene rearrangements with multiple different partners (Fig. 3).
• As in spindle cell lipoma, RB1 loss is a defining feature in both cellular angiofibroma and myofibroblastoma. These tumors have some overlapping histologic and immunophenotypic features in addition to the same genetics, so distinction relies on a combination of clinical history and pathologic features.
• Translocation-associated fibroblastic neoplasms are typically benign to low-grade neoplasms with monotonous cytology and characteristic architectural features. An NGS panel can confirm the diagnosis, though this is often not required because the morphology is typical and molecular analogue immunohistochemical (IHC) stains exist for many of these tumors with relatively high sensitivity and specificity.
- Solitary fibrous tumor: NAB2::STAT6 fusion; prominent collagenous background and staghorn vasculature.
- Dermatofibrosarcoma protuberans: COL1A1::PDGFB fusion; dermal tumor with diffuse storiform architecture and honeycomb/septal infiltration into fat. Fibrosarcomatous transformation can mask the storiform architecture and gives the tumor metastatic potential.
- Inflammatory myofibroblastic tumor: ALK rearrangement; bland myofibroblastic proliferation with prominent mixed inflammatory infiltrate. Malignant versions exist, often with more epithelioid morphology.
- Low-grade fibromyxoid sarcoma/sclerosing epithelioid fibrosarcoma: FUS or EWSR1 rearrangement, usually with CREB3L2 or CREB3L1; cellular spindle cell neoplasm with mixed collagenous and myxoid features (low-grade fibromyxoid sarcoma) or sclerotic tumor with bland epithelioid cytology (sclerosing epithelioid fibrosarcoma). Additional fusions involving KMT2A and YAP1 that show similar morphological features to sclerosing epithelioid fibrosarcoma have been found.
- EWSR1::SMAD3-positive fibroblastic tumor: EWSR1::SMAD3 fusion; newly described fascicular, bland spindle cell proliferation with distinct zonation (hypocellular central region and increased peripheral cellularity) with ERG IHC positivity.
- Superficial CD34-positive fibroblastic tumor: PRDM10 rearrangements; low-grade fascicular proliferation of spindle cells with marked nuclear pleomorphism.
Vascular
• Fusion testing is not required but helpful in the diagnosis, particularly in the case of the low-grade malignancies and hemangioendotheliomas.
• Epithelioid hemangioma is defined by recurrent fusions involving FOS and FOSB. Fusion testing is not required but can be useful in more cellular cases with atypical cytologic features.
• Epithelioid hemangioendothelioma: WWTR1::CAMTA1 fusion or rarely TFE3::YAP1 fusion; these malignant tumors show more primitive vascular differentiation, epithelioid morphology, intracytoplasmic lumens, and a distinct myxohyaline stroma that distinguishes them from epithelioid angiosarcoma. The CAMTA1 fusion or IHC stain may be used to confirm the diagnosis.
• Pseudomyogenic hemangioendothelioma: FOSB gene fusions, usually with SERPINE1 or ACTB; histologically, shows a rhabdomyoblastic-like appearance but stains for keratins and vascular markers.
• Angiosarcoma, like other high-grade sarcomas, shows complex genomic changes. The presence of MYC gene amplifications (tested by IHC, FISH, or NGS) is seen in most cases of postirradiation or lymphedema-associated angiosarcoma. This finding is very useful in distinguishing post-irradiation atypical vascular lesions from true angiosarcoma.
Pericytic and smooth muscle
• While the diagnosis of glomus tumors is generally based on morphology, examples of deep, gastrointestinal, or malignant glomus tumors may confound the diagnosis. Most glomus tumors (including malignant variants) show recurrent fusions involving the NOTCH family of genes, most commonly MIR143::NOTCH1/2/3.
• Distinct from classical leiomyosarcoma, inflammatory leiomyosarcoma is a low-grade malignancy that has been shown to have a recurrent karyotypic pattern that is best seen on mRNA microarray technology, such as OncoScan™. These neoplasms show a near-haploid genotype that is thought to be a relevant driver of the disease process. Some tumors may show whole genome duplication afterwards, resulting in a pseudo-hyperdiploid karyotype that may signal transformation to a higher-grade malignancy. Recent studies have demonstrated a number of cases with rhabdomyoblastic IHC staining, and new terminology (inflammatory rhabdomyoblastic tumor) has been suggested.
Skeletal muscle
• Molecular testing in rhabdomyoblastic tumors is best utilized in the differentiation of embryonal and alveolar rhabdomyosarcoma. Both tumors may have a solid small round blue cell morphology and skeletal muscle staining with IHC. Myogenin IHC tends to be more diffuse in alveolar rhabdomyosarcoma; however, definitive diagnosis requires molecular detection of the typical FOXO1 fusion with either PAX3 or PAX7 in alveolar rhabdomyosarcoma. Embryonal rhabdomyosarcoma may show recurrent mutations in the RAS family of genes or DICER1 in some syndromic patients. The distinction between the alveolar and embryonal subtypes is necessary because of the variation in prognosis and treatment.
• Spindle cell rhabdomyosarcoma falls into three distinct molecular groupings. Congenital and infantile tumors are most often translocation-driven, with fusions involving VGLL2, SRF, READ1, NCOA2, and CITED2. Another subset of tumors in adolescents and young adults shows mutations in the MYOD1 gene. The third category does not have recurrent genetic abnormalities. Additionally, some intra-osseous variants may show EWSR1, FUS, or MEIS1::NCOA2 rearrangements.
Gastrointestinal stromal tumor (GIST)
• All GISTs should be tested for mutational status, as these mutations predict response to treatment and prognosis.
• Mutations most commonly occur in KIT (exons 9, 11, 13, 14, or 17) and second most commonly in PDGFRA (exons 12, 14, and 18).
• Immunostaining for cKIT (CD117) does not imply the presence of a KIT mutation.
• SDH-deficient GISTs are negative for KIT and PDGFRA mutations and show mutations in SDHA, SDHB, SDHC, or SDHD. These mutations are typically screened by assessing for loss of SDHB IHC staining, which picks up mutations in any of the four genes. SDH-deficient GISTs are more common in younger patients in the stomach.
• Some GISTs will instead show mutations in RAS family genes.
• A small proportion of GISTs may be negative for all four of these mutations, called quadruple wild-type GIST.
Uncertain differentiation and round cell tumors
• Undifferentiated round to spindle cell tumors and those with monomorphic cytology should be considered for large panel NGS fusion testing. IHC has been found to show much overlap in this category of tumors, and NGS testing offers the ability to pick up non-classical examples as well as discover new fusion partners.
• Currently, Ewing and Ewing-like round cell sarcomas can be split into six overall categories (Table 1). New fusion partners continue to be discovered, and the particular fusion may affect treatment and prognosis.
Certain neoplasms show bi-immunophenotypic staining patterns that may lead to confusion. Molecular testing can be confirmatory.
- Angiomatoid fibrous histiocytoma: EWSR1::ATF1, FUS::ATF1, or EWSR1::CREB1; a low-grade malignancy with EMA and desmin co-positivity and prominent lymphoid cuffing.
- Ossifying fibromyxoid tumor: Most commonly PHF1 rearrangements (50% of cases), rarely rearrangements in BCOR or SUZ12, suggesting a genetic overlap with endometrial stromal sarcoma; low-grade malignancy with prominent peritumoral metaplastic bone formation and often co-positivity for keratins, S100, or desmin.
- Myoepithelial neoplasms of soft tissue: EWSR1 or FUS rearrangements with several different partners; keratin and S100 co-positivity with myxoid to myxocollagenous background and bland round to spindled cells. INI1 is lost in a subset. These tumors show different genetics to salivary gland myoepithelial neoplasms, which are often governed by PLAG1 rearrangements.
- Extraskeletal myxoid chondrosarcoma: NR4A3 rearrangement, most commonly with EWSR1. Keratin, S100, neuroendocrine, or myoepithelial markers may be nonspecifically positive. Morphologic and immunophenotypic overlap with myoepithelial neoplasms may cause diagnostic difficulty, and because of the presence of EWSR1 as a partner in both, NGS testing is recommended to assess the partner gene to distinguish these two.
• Phosphaturic mesenchymal tumor: in patients with clinical evidence of hypophosphatemia and/or osteomalacia, serum testing may be performed for increased FGF23 secretion. Most of these tumors show fusions involving FN1::FGFR1 or FN1::FGF1.
• NTRK gene rearrangements have been seen in an increasing spectrum of mesenchymal tumors. The prototypical infantile fibrosarcoma is defined by ETV6::NTRK3. However, NTRK fusions have now been seen in cellular mesoblastic nephroma as well as various myxoid soft tissue tumors with bland cytology and possible co-positivity for S100 and CD34, including lipofibromatosis-like neural tumor. Some uterine and soft tissue neoplasms with fibrosarcoma-like morphology also define a new subset of NTRK-rearranged sarcomas.
• A subset of PEComa is driven by rearrangements in TFE3; often not required for the diagnosis, as the combination of myogenic and melanocytic markers is specific enough in most instances to diagnose PEComa.
• Recent studies have shown that the majority of true intimal sarcomas show amplifications in MDM2, similar to well/dedifferentiated liposarcoma. Intimal sarcoma may have variable differentiation and immunostaining. The presence of a luminal mass in the pulmonary or cardiac vasculature should prompt FISH testing for MDM2 to confirm the diagnosis.
Bone and cartilage
• The diagnosis of primary bone lesions is still based most heavily on the morphology coupled with the radiological imaging.
• Some distinct exceptions where molecular testing can be diagnostically useful:
- Aneurysmal bone cyst: USP6 gene rearrangements; cystic, giant cell-rich neoplasm with reactive woven bone formation.
- Low-grade central osteosarcoma/parosteal osteosarcoma: MDM2 amplifications; low-grade osteoblastic tumors that, similar to well-differentiated liposarcoma, have potential to dedifferentiate. MDM2 testing by FISH can help to distinguish from reactive or benign bone-forming tumors.
- Giant cell tumor of bone/chondroblastoma: both show unique and specific mutations in H3F3A or H3F3B. IHC testing is useful as a molecular adjunct.
- Conventional chondrosarcoma: IDH1 or IDH2 mutations in a subset of chondrosarcoma; can be diagnostically useful in small biopsies and dedifferentiated examples.
- Mesenchymal chondrosarcoma: HEY1::NCOA2 fusion; small round blue cell sarcoma with cartilaginous maturation.
Meet the Authors
Dr. Obeidin has been an author for PathologyOutlines since 2018 and part of the PathologyOutlines editorial board since January 2022. He is currently an Assistant Professor of Pathology at Northwestern University Feinberg School of Medicine. He obtained his M.D. at the Medical College of Georgia and then completed his Anatomic and Clinical Pathology residency at Northwestern University. He then completed a fellowship in General Surgical Pathology and Bone and Soft Tissue Pathology at the University of California, Los Angeles.

 XML Download
XML Download