

 PDF
PDF Citation
Citation Print
Print



I. Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children and young adults; it represents 3% of childhood malignancies and is frequently found in patients around 14 years of age1. Thirty percent of cases appear in the head and neck region. RMS is histologically classified as embryonal, alveolar, pleomorphic, or spindle cell/sclerosing according to the World Health Organization. Its treatment depends on age at diagnosis, tumor size, and areas involved and normally involves surgery, chemotherapy, and radiotherapy2.
The head and neck are the most common RMS locations. Within this region, RMS has been identified in the parameningeal, non-parameningeal, and orbital areas3. Due to the use of multimodal therapy (surgery, chemotherapy, and radiation therapy), the cure rate in children has improved to greater than 70%; however, there is a poor prognosis for advanced cases, with a 5-year survival rate of only 30%-50%. This prognosis is improved in children ages 1-9 years but is worse in those older than 10 years and those presenting with metastasis2.
Even though head and neck rhabdomyosarcoma (HNRMS) presents aggressively, there are few articles referring to its clinical pathology. Financial limitations such as molecular testing, especially in those with embryonal components, impede the availability of knowledge regarding this topic4. This rare malignancy is commonly misdiagnosed, and a small number of patients is normally included in related studies5. There are no recent systematic reviews on this matter in digital databases, and most literature articles do not report immunohistochemical analyses.
Because of this lack of information regarding HNRMS, the objective of this study was to perform a systematic review of its clinical-pathological profile and most relevant prognostic factors in pediatric patients.
II. Materials and Methods
This systematic review was conducted to determine the clinical-pathological profile and relevant prognostic factors of HNRMS in pediatric patients. A systematic literature review was conducted following the guidelines of the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines. This systematic review was recorded in the PROSPERO database under the number CRD42020207099 and is available at PROSPERO (https://www.york.ac.uk/).
The data were collected from the electronic databases PubMed, Lilacs, Embase, Scopus, and Web of Science. The search strategy used Boolean operators (AND and OR): [ALL (“Rhabdomyosarcoma”) AND (pediatric OR child OR children OR young OR adolescent OR hebiatric) AND (head and neck)]. The End-Note reference manager was used to save search records and eliminate duplicates.
The studies were selected by two reviewers independently. In the first stage, the titles and abstracts were read; in the second stage, the complete texts were read to identify articles that met the eligibility criteria. The inclusion criteria were as follows: patients diagnosed with rhabdomyosarcoma between 0 and 18 years of age, case series studies with a minimum of five cases, case-control studies, randomized clinical studies, retrospective clinical studies, and studies published in English, Portuguese, or Spanish. Articles were excluded if the reported cases involved malignant tumors other than rhabdomyosarcoma, patients older than 18 years, clinical cases, or literature reviews.
The following data were collected: first author, year, country of study, type of study, patient sex, patient age, tumor location, tumor classification, tumor stage, histopathological analysis, immunohistochemical analysis, treatment, follow-up, mean survival, and outcome. We included articles that contained at least 60% of the data in the review. During each stage of the study selection, a third reviewer helped resolve discrepancies.
The quality of studies was assessed using the STROBE (Strengthening the Reporting of Observational Studies) checklist.
III. Results
1. Study selection
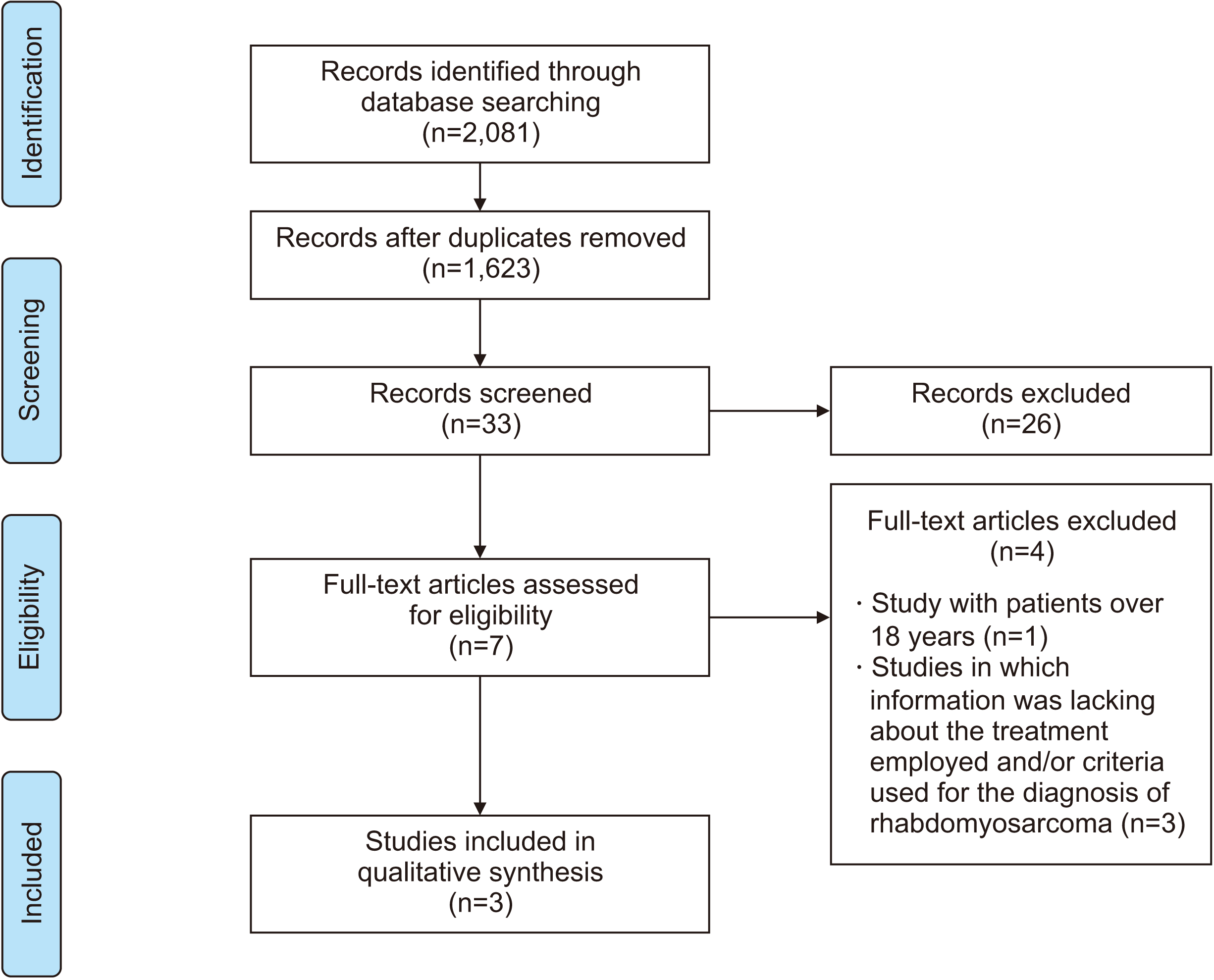
The initial database search resulted in 2,081 articles. After removing duplications, 1,623 articles remained. After reading the titles and abstracts, we assessed 33 articles. A flowchart describing the study selection phase is shown in Fig. 1.
2. Study characteristics
The three selected studies were observational; published in English in 19866, 20153, and 20184; and conducted in Germany, Japan, and India, respectively. A total of 380 patients was included in the meta-analysis study. The mean age of the patients was 13 years 6 months, and 67.4% (256 patients) of the patients were male. Of the included patients, 159 were diagnosed with embryonal type, 141 were diagnosed with alveolar type, 71 were diagnosed with spindle cell/sclerosing type, and 9 were diagnosed with pleomorphic type. Diagnoses were based on histopathology and immunohistochemistry in all included studies. Treatment protocols consisted of chemotherapy and radiotherapy, which were usually administered in combination with surgery. The follow-up period was at least 25 months in all studies. The mean survival rate was greater than 50%3,4,6. Detailed information and data on follow-up, relapse, mean survival, and outcomes are shown in Table 1.
3. Risk of bias within studies
All studies achieved at least 15 points on the STROBE checklist, which indicates that they are good studies.(Table 2)
4. Results of individual studies
Schmidt et al.6 suggested division of embryonic RMS into three subtypes of primitive with predominantly undifferentiated cells (<10% rhabdomyoblasts), intermediate with 10%-50% rhabdomyoblasts, and well-differentiated with more than 50% rhabdomyoblasts. The study did not show any significant differences between subtypes, although they did show different body locations of origin. The primitive and intermediate subtypes occurred predominantly in the head and neck regions, while the well-differentiated subtype was most commonly recurrence at other sites. Expression of vimentin and desmin was positive in all three subtypes, with vimentin being the most significant in the primitive embryonic rhabdomyosarcoma subtype. Myoglobin expression was positive only in the well-differentiated embryonic rhabdomyosarcoma subtype. In addition, the primitive and intermediate embryonic rhabdomyosarcoma subtypes were usually diagnosed earlier than the well-differentiated type. At the time of diagnosis, intermediate embryonic rhabdomyosarcoma presents a better response to chemotherapy compared to the well-differentiated subtype.
Yasui et al.3 presented 16 cases of embryonic spindle/sclerosing cell RMS and 10 cases of primary tumors in the head and neck region. Regarding immunohistochemistry, the study showed that expression of focal myogenin (10%) and diffuse MYOD1 (>50%), associated with the discrete presence of desmin, can confirm diagnosis. The results suggest that fusiform/sclerosing RMS in pediatric patients has a worse prognosis than embryonic RMS since more than half of these tumors experience local or distant recurrence. The data obtained were not subjected to statistical analysis because of the small sample size.
Rekhi et al.4 performed a study involving 300 patients diagnosed with RMS. Regarding location, 42% occurred in the head and neck region, with a higher frequency of alveolar RMS. Among patients younger than 20 years, the embryonic type was most frequent. The study established a correlation between inhibition of markers, such as desmin, myogenin, and MYOD1, and RMS subtype. Myogenin expression is strongly related to the alveolar type. The expression of MYOD1 correlated with the predominance of spindle/sclerosing cell type. Tumor sizes smaller than 5 cm and absence of metastasis significantly contributed to longer disease-free and overall survival.
IV. Discussion
Previous studies have reported 5% of malignant tumors in children in the head and neck region7,8. This value was updated in 2020, when RMS represented 3% of malignant tumors in children1.
1. Epidemiology
The total number of subjects observed in the selected studies was 380, of which 256 were male (67.4%) and 124 were female (32.6%), with a male/female ratio of 2:13,4,6. These values are comparable with those observed individually in each study included in this review, as reported by Lyos et al.9. The mean age of included patients was 13 years 6 months3,4,6. This is different from other studies, possibly because approximately 21% of cases reported in the included studies were sclerosing and pleomorphic RMS variants with a greater prevalence in adults10.
Among the types of RMS, embryonic was the most prevalent, comprising 159 diagnosed cases (41.8%). Alveolar type was the second most common, with 141 cases (37.1%), followed by 71 cases of sclerosing RMS (18.7%) and 9 cases of pleomorphic RMS (2.4%)3,4,6.
For location, soft tissues were most commonly affected, observed in 170 cases (44.7%). Sixty-three cases (16.6%) involved the extremities, while the head and neck region was affected in 157 cases (41.3%)3,4,6. These values are proportional to those observed by Dillon et al.11 and Radzikowska et al.12.
2. Diagnosis
Tumors can be identified by computed tomography or magnetic resonance imaging, and diagnoses can be confirmed by biopsy of altered and healthy cells. Immunohistochemistry is useful in determining the subtypes of RMS and choosing treatment3,4,6,13. Embryonic and alveolar RMS were the two main histological subtypes observed3,4,13.
Markers of desmin, myogenin, and MYOD1 expression may be viable indicators of tumor subtype. In the study by Rekhi et al.4, desmin had a positive expression greater than 90% (292/299), myogenin was observed in >70%, and MYOD1 in 65.3% of myoglobin. MYOD1 is the most indicated marker for reliable diagnosis in cases of sclerotic and spindle cell RMS3,4. However, use of myogenin alone can lead to misdiagnosis, as its expression also can be related to alveolar RMS, which was reported in 72.3% (34/47) of cases in the study by Rekhi et al.4.
3. Etiopathogenesis, treatment, and prognosis
Schmidt et al.6 proposed the establishment of embryonic RMS subtypes based on the correlation between presence and predominance of rhabdomyoblasts. However, the results presented did not show significant differences based on this criterion. The presence and proportion of these cells in immunohistochemical studies showed no correlation with clinicopathological features3.
The head and neck region is the most common anatomical site for sclerosing and fusiform RMS. MYOD1 mutations have been reported in 3 of the 13 investigated cases of sclerosing-type RMS14. Another study identified the MYOD1 mutation in 30 cases of sclerosing and/or spindle cell RMS in patients aged 2-94 years, including 15 children15.
The sclerosing type of RMS is commonly diagnosed at an advanced stage because of its primary site (or extremities) and tumor size at initial diagnosis3. Alveolar RMS also showed a significant correlation with metastasis16, and patients with tumors <5 cm and those free from metastasis had better overall survival and disease-free survival4.
Surgery has demonstrated that local metastasis is a possibility, albeit rarely3. When possible, the suggested treatment involves complete tumor resection in addition to adjuvant therapy (chemotherapy and radiotherapy) to reduce the chances of local and distant metastases3,4. When the MYOD1 mutation is present, 83% of pediatric patients died from the disease, even with multimodal treatment15.
V. Conclusion
There are few complete studies on RMS. This review allowed us to determine that RMS has a 2:1 male/female ratio, a mean age of 13 years six months, and a high prevalence of embryonic type. For diagnostic measures, use of the MYOD1 marker is reliable in cases of sclerotic and spindle cell RMS. The presence of this marker may indicate a poor prognosis. On the other hand, patients diagnosed with tumors smaller than 5 cm who did not experience metastasis had a favorable prognosis when treated with complete resection followed by chemotherapy and/or radiotherapy.
 XML Download
XML Download