

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
In the 1990s to early 2000s, eight non-cardiac drugs were withdrawn from the market because of their association with Torsade de Pointes (TdP), a potentially life-threatening ventricular arrhythmia condition [1]. Up to 90% of new compounds that pass preclinical testing fail at the clinical trial phase, with cardiotoxicity accounting for the failure of 45% of the compounds [2]. Hence, the International Conference for Harmonization (ICH) presented the regulatory guidelines S7B and E14 in 2005, which focuses on two markers to assess TdP risk: in vitro inhibition of a single human Ether-à-go-go-Related Gene (hERG) potassium channel (representing the rapidly activating delayed rectifier potassium current, or IKr) and prolongation of the heart rate corrected QT (QTc) interval in clinical studies [3,4]. However, hERG is not the only cardiac ion channel that regulates ventricular repolarization, and prolongation of the QTc interval alone is insufficient to be used as a predictor of clinical TdP risk [5]. Therefore, to address these problems, the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative was proposed jointly by the US Food and Drug Administration, the Cardiac Safety Research Consortium, and the Health and Environmental Sciences Institute in July 2013 [6].
The three preclinical elements of the CiPA initiative include (1) in vitro multiple cardiac ion channel effects of a drug (hERG/IKr, Cav1.2/ICa, and Nav1.5/INa) in heterologous expression systems, (2) the integration of drug/channel interaction data from in silico models of human ventricular electrophysiology to evaluate and predict changes in the human action potential (AP), and (3) in vitro evaluation of drug effects in a cardiomyocyte assay involving human-induced pluripotent stem cell-derived cardiomyocytes (human iPSC-CMs) and comparison with in silico results [7]. In particular, human iPSC-CMs can confirm changes in human AP and repolarization, irrespective of the cellular mechanism directly affecting the human cardiac ion channel or being difficult to predict using an in silico reconstruction model. However, the current CiPA initiative is the first platform, and the in vitro multiple cardiac ion channel assay is only dependent on the artificial expression of a selective cardiac ion channel in human embryonic kidney or Chinese hamster ovary cell lines, based on the two ICH guidelines S7B and E14. The sole reliance on simplified mammalian model cell lines possessing interspecies differences in biological pathways, ion channels, and pharmacokinetic properties puts human lives at risk. Thus, developing an in vitro multiple ion channel screening system based on human iPSC-CMs could provide more relevant and reproducible results for drug safety assessment.
In this study, we established a modified human iPSC-CM-based in vitro multiple ion channel screening system that can analyze changes in electrophysiological functions in accordance with the CiPA concept.
METHODS
Chemicals
Sotalol, chlorpromazine, mexiletine, E-4031, nifedipine, tetrodotoxin (TTX), and 4-aminopyridine were purchased from Tocris Cookson and dissolved in distilled water. Fibronectin was purchased from Gibco.
Cell culture
Human iPSC-CMs (Cardiosight-S C-001; NEXEL) were cultured for single-cell electrophysiological experiments. Frozen vials of human iPSC-CMs were thawed quickly in a water bath at 37°C and mixed with an ice-cold plating medium (Cardiosight-S Advanced Plating Medium CM-010A). The cells were transferred to 24-well culture plates (1.4 × 105 cells/well) containing 50 µg/ml fibronectin-coated glass coverslips at a low density to yield single uncoupled cells. Thereafter, the cells were maintained in a humidified incubation chamber containing 5% CO2 at 37°C. After 24 h of incubation, the plating medium was replaced with a maintenance medium (Cardiosight-S Advanced Maintenance Medium CM-001A), which was changed every 2 days. The cells were cultured for 1 week and used 7–14 days post-thaw for electrophysiological analysis.
Electrophysiological analysis
To record whole-cell membrane currents, human iPSC-CMs were placed in an experimental chamber (RC-26G; Warner Instruments) mounted on the stage of an Olympus BX51WI upright microscope. A heated external solution (37°C) was continuously perfused into the chamber. The patch pipettes were pulled from borosilicate glass capillaries (TW100-4; World Precision Instruments) using a vertical puller (PP-830; Narishige Scientific Instrument Laboratory) to establish 1–3 MΩ when filled with the internal solution. We recorded whole-cell membrane voltage or current using a patch clamp amplifier (Axopatch 200 B; Axon Instruments) in voltage-clamp or current-clamp mode and an A/D converter (Axon Digidata 1550B; Axon Instruments) data acquisition system controlled by software (pCLAMP 10; Axon Instruments).
The external solution for APs and IKr channel recordings contained NaCl (150 mM), KCl (5.5 mM), HEPES (10 mM), NaH2PO4 (0.33 mM), CaCl2 (1.8 mM), MgCl2 (1 mM), and dextrose (10 mM), with the pH adjusted to 7.4 using 1 M NaOH. The internal solution for APs and IKr channel recordings contained K-Asp (120 mM), NaCl (5 mM), KCl (20 mM), HEPES (10 mM), EGTA (5 mM), Mg-ATP (5 mM), and CaCl2 (1.8 mM), with the pH adjusted to 7.25 using 1 M KOH. The external solution for ICa channel recordings contained NaCl (135 mM), CsCl (10 mM), HEPES (5 mM), NaH2PO4 (0.33 mM), CaCl2 (1.8 mM), MgCl2 (0.5 mM), and dextrose (16.6 mM), with the pH adjusted to 7.4 using 1 M NaOH. The external solution for INa channel recordings contained NaCl (130 mM), CsCl (15 mM), HEPES (5 mM), NaH2PO4 (0.33 mM), CaCl2 (1.8 mM), MgCl2 (0.5 mM), and dextrose (16.6 mM), with the pH adjusted to 7.4 using 1 M NaOH. The internal solution for ICa and INa channel recordings contained Cs-Asp (120 mM), NaCl (5 mM), CsCl (20 mM), TEA-Cl (10 mM), HEPES (10 mM), EGTA (10 mM), and Mg-ATP (5 mM), with the pH adjusted to 7.25 using 1 M CsOH.
Statistical analyses
Data acquisition and statistical analyses were performed using pCLAMP (Axon Instruments) and Origin 8.0 (OriginLab Corp.). Data are presented as the mean ± SEM, and n represents the number of individual whole-cell human iPSC-CM recordings. Student’s t-test was used to test for statistical significance as appropriate. Statistical significance was set at p < 0.05.
RESULTS
Electrophysiological characterization of human iPSC-CMs
Using the whole-cell patch clamp technique in the current-clamp mode, we first measured the APs in spontaneously beating cells isolated from human iPSC-CMs. Based on the distinct heterogeneity of human iPSC-CMs, three major AP subtypes (nodal, atrial, or ventricular-like) (Fig. 1 and Table 1) were characterized based on a predominant AP morphology. Fig. 1A shows a schematic overview of representative traces of spontaneous APs recorded from human iPSC-CMs. Differentiation of ventricular-like APs (Fig. 1A, bottom) was performed on the relatively more negative resting membrane potential (RMP) and rapid AP upstroke with a long plateau phase. Atrial-like APs showed a lack of a plateau phase and shorter AP duration compared to those shown by ventricular-like APs (Fig. 1A, middle). A prominent phase 4 depolarization with a less negative RMP than those of atrial- and ventricular-like CMs was characteristic of nodal-like APs, resulting in slower AP upstroke (Fig. 1A, top). Most cells (~87.5%) showed ventricular-like APs, whereas nodal-like APs and atrial-like APs were also recorded among all the cells (n = 80, Fig. 1B). Fig. 1C summarizes the AP characteristics of the human iPSC-CMs. The RMP, total amplitude (TA), maximum upstroke velocity (dV/dtmax), and the AP duration at 90% (APD90) repolarization were analyzed. The mean APD90 of the ventricular-like CMs was 730.61 ± 1.01 ms, and all 70 cells had an APD90 > 600 ms; hence, only cells with an APD90 > 600 ms were used for testing the cardiotoxicity of drugs. Atrial-like CMs had a control APD90 between 500 and 600 ms (n = 8, with a mean APD90 of 541.63 ± 3.54 ms), and all two cells classified as nodal-like CMs were distinguished from atrial-like APs by a smaller TA and a significantly lower dV/dtmax. Table 1 summarizes the AP characteristics of human iPSC-CMs and native human ventricular cardiomyocytes (native human VCMs). The RMP of ventricular-like APs in human iPSC-CMs is less negative, with values ranging from −62 to −65 mV compared with the AP parameters of native human VCMs [8]. The TA of ventricular-like APs (119–122 mV) was comparable to that of native human VCMs (105–108 mV) despite the depolarized RMP in human iPSC-CMs. The dV/dtmax of ventricular-like APs in human iPSC-CMs ranged from 45 to 48 V/s, which was slower than that of native human VCMs (182–248 V/s). The APD90 of ventricular-like APs (730–732 ms) in human iPSC-CMs was longer than that of native human VCMs (337–365 ms). Although some differences were observed in some of the AP parameters between human iPSC-CMs and native human VCMs, most of the AP parameters recorded in human iPSC-CMs are distinct from human cardiac cells. Therefore, we suggest that human iPSC-CMs have the potential to serve as a new human-based model for predicting drug-induced cardiotoxicity and could replace native human cardiomyocytes.
Effects of selective cardiac ion channel inhibitors on APs in human iPSC-CMs
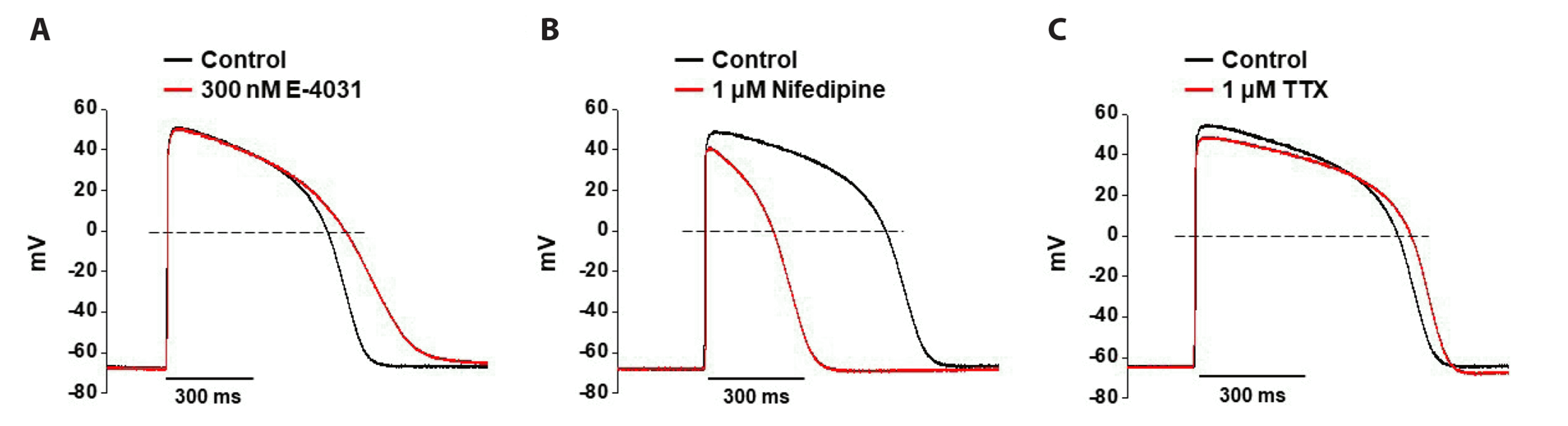
To identify the utility of human iPSC-CMs as testbeds for drug-induced cardiotoxicity, we investigated whether the APs were actually affected by cardiac ion channel inhibitors, such as E-4031, nifedipine, and TTX specific for IKr, ICa, and INa channels, respectively (Fig. 2). Application of 300 nM E-4031 induced a significant prolongation of the APs; APD90 was increased by 20.6 ± 2.12%. There were no significant changes in the RMP, TA, and dV/dtmax (n = 3, Fig. 2A). Nifedipine significantly shortened the APD90 to 51.92 ± 1.2% at 1 µM, with minor reductions in TA (n = 3, Fig. 2B). Similar to nifedipine, 1 µM TTX had no significant effect on AP parameters, except for a minor reduction in APD90 and TA (n = 3, Fig. 2C). These results demonstrate that human iPSC-CMs effectively summarize the electrophysiological behavior of native human ventricular CMs and are suitable for cardiac-related drug safety evaluation.
Effects of the high, intermediate, and low TdP risk drugs on APs in human iPSC-CMs
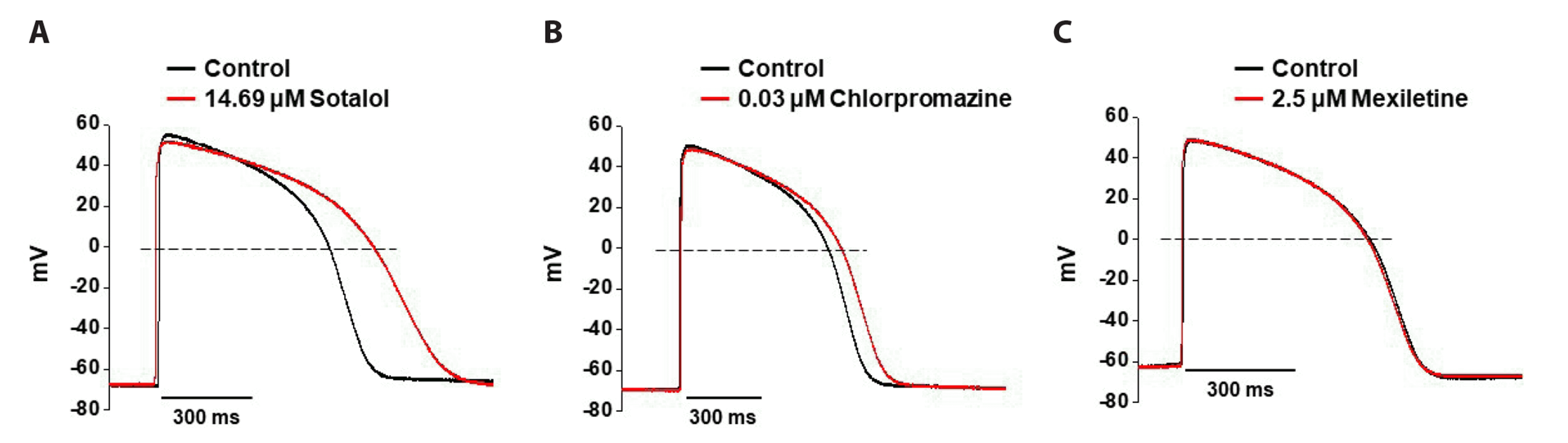
In the CiPA initiative, human iPSC-CM-based cardiovascular safety assessment and pharmacology tests were performed using various drugs with clinical torsogenic information [9]. We first tested the effects of high, intermediate, and low TdP risk drugs on the AP waveforms of human iPSC-CMs using whole-cell patch-clamp techniques. Sotalol, a high TdP risk drug, significantly prolonged the APD90 to 26.12 ± 1.2% at 1× Cmax (14.69 µM), with a minor reduction in TA (n = 3, Fig. 3A). There were no significant changes in the RMP and dV/dtmax of APs after exposure to sotalol. Chlorpromazine, an intermediate TdP risk drug, also increased APD90 by 8.79 ± 2.1% at 1× Cmax (0.03 µM); however, it did not induce significant changes in RMP, TA, or dV/dtmax (n = 3, Fig. 3B). Mexiletine, a low TdP risk drug, at 1× Cmax (2.5 µM) slightly decreased APD90 by 1.26 ± 1.5% without changing other AP parameters (n = 3, Fig. 3C).
Effects of the high, intermediate, and low TdP risk drugs on cardiac ion channel currents in human iPSC-CMs
Repolarization-related currents (IKr) and depolarization-related currents (ICa and INa) are the major cardiac ion currents most closely related to the cardiac safety of drugs [10]. To understand the cardiotoxic mechanism of TdP risk drug-induced modification of AP waveforms, the effects of these drugs on IKr, ICa, and INa in human iPSC-CMs were analyzed using the whole-cell patch clamp technique. Each ion current was activated using appropriate one-step pulse protocols under pharmacological conditions to selectively isolate the currents (see the Materials and Methods section).
To investigate the effects of the high, intermediate, and low TdP risk drugs on the repolarization-related current, IKr was recorded. To record IKr, human iPSC-CMs were depolarized for 2 sec to +20 mV from a holding potential of –80 mV, followed by repolarization back to –40 mV for 2 sec. Fig. 4 shows the concentration-dependent effects of sotalol, chlorpromazine, and mexiletine on IKr amplitude in human iPSC-CMs. Sotalol at 1×, 3×, and 10× free plasma Cmax reduced the amplitude of IKr by 12.07%, 33.68%, and 71.34%, respectively (n = 4, Fig. 4A, B). Chlorpromazine at the same multiples of free plasma Cmax attenuated the amplitude of IKr by 9.73%, 12.16%, and 18.88%, respectively (n = 4, Fig. 4C, D). However, mexiletine exhibited only a weak inhibitory effect on IKr. Mexiletine reduced the amplitude of IKr by 1.26%, 2.75%, and 9.53% at 1×, 3×, and 10× free plasma Cmax, respectively (n = 4, Fig. 4E, F).
The effects of high, intermediate, and low TdP risk drugs on depolarization-related currents, ICa and INa, were analyzed. The ICa of human iPSC-CMs was elicited using a one-step pulse from a holding voltage of –50 mV to 0 mV for a duration of 300 ms. Fig. 5 shows the representative ICa traces under control conditions and after exposure to sotalol, chlorpromazine, and mexiletine. All drugs were found to reduce the amplitude of ICa in a concentration-dependent manner, with mexiletine having the greatest effect and sotalol having the least effect. Sotalol at 1×, 3×, and 10× free plasma Cmax reduced the amplitude of ICa by 7.91%, 18.91%, and 22.25%, respectively (n = 4, Fig. 5A, B). Chlorpromazine at the same multiples of free plasma Cmax attenuated the amplitude of ICa by 17.39%, 37.92%, and 63.04%, respectively (n = 3, Fig. 5C, D). Additionally, mexiletine had the most potent inhibitory effect on ICa. Mexiletine reduced the amplitude of ICa by 32.6%, 43.59%, and 53.11% at 1×, 3×, and 10× free plasma Cmax, respectively (n = 4, Fig. 5E, F). In human iPSC-CMs, INa was generated using a step pulse from a holding voltage of –100 mV to –40 mV for a duration of 50 ms. All three drugs also inhibited INa in the micromolar range (Fig. 6A, C, and E). Sotalol at 1× free plasma, Cmax significantly reduced the amplitude of INa by 19.23%, whereas chlorpromazine and mexiletine reduced the amplitude of INa by approximately 2.8% and 5.32%, respectively, at 1× free plasma Cmax. These results, together with those for APs, showed that our data are consistent with the electrophysiological mechanism by which each drug acts on cardiac ion channels and AP waveforms.
DISCUSSION
The aim of the present study was to establish a new in vitro human stem cell-based multiple cardiac ion channel assay to assess drug safety in human iPSC-CMs. Here, we assessed the potential of human iPSC-CMs as a new alternative in vitro multiple-ion channel assay model for testing putative arrhythmogenic drugs. Our studies showed that human iPSC-CMs effectively recapitulated the electrophysiological behavior of each cardiac ion channel inhibitor, confirming the plausibility of a system for preclinical drug safety pharmacological assessment. In comparison with the heterologous expression system, the existing preclinical in vitro multiple ion channel assay demonstrated a reasonable correlation, thereby indicating the potential use of human iPSC-CMs for electrophysiological cardiac safety screening.
Cardiotoxicity screening, including in vitro multiple ion channel assays and in vivo QT assays, is an important element in preclinical drug safety evaluations prior to first-in-human clinical studies. The previous safety screening platform was based primarily on the predictive link between drug-induced in vitro hERG inhibition and in vivo/clinical QT interval prolongation and TdP; however, several drugs that have passed laboratory animal tests show unanticipated fatal cardiotoxicity during the clinical phases [11]. Furthermore, these platforms have limitations regarding species differences, ethical concerns, and cost [12]. Therefore, human stem cell-based platforms have attracted attention because they can overcome the limitations of currently used platforms for the preclinical safety of pharmaceutical chemicals. Our present study supports that human iPSC-CMs are an attractive platform for cardiotoxicity screening, as they express the major cardiac ion channels and recapitulate spontaneous electrical and mechanical activities, similar to isolated human cardiomyocytes. 1) All AP parameters from human iPSC-CMs were similar to published values in human cardiomyocytes [8]. 2) Human iPSC-CMs responded to drug application in a manner similar to isolated human cardiomyocytes [13]. Nifedipine resulted in AP shortening, whereas E-4031 and TTX prolonged the AP duration. Thus, we concluded that human iPSC-CMs can recapitulate cardiotoxicity and identify the effects of well-characterized compounds.
Previous studies have demonstrated that sotalol, chlorpromazine, and mexiletine inhibit hERG currents, which may explain the cardiac toxicity of these drugs [14-16]. Although it is widely accepted that TdP, known as fatal ventricular arrhythmia, is primarily caused by hERG inhibition, the limitations of such a simple interpretation have also been reported [17]. An abnormally prolonged QT is sensitive; however, it is not highly specific for predicting which drugs can cause TdP. Additionally, drug-induced blockade of hERG does not indicate a clear correlation with QT interval prolongation risk or the occurrence of proarrhythmia. Therefore, the integrated effects of the non-hERG cardiac ionic currents should be considered. Inhibition of both INa-late and ICa has been associated with a reduction in QT interval prolongation and TdP, even in the presence of hERG inhibition [18-20]. Therefore, when assessing TdP liabilities, a new alternative in vitro model and an integrative interpretation are needed to understand the cardiotoxicity effects of drugs, affecting multiple types of cardiac ion channels beyond hERG.
In this study, we investigated the effects of sotalol, chlorpromazine, and mexiletine on AP features and major cardiac ion channels, including IKr, ICa, and INa channels, in human iPSC-CMs. Included in the drugs tested was one representative drug from each of the CiPA risk categories (high-, intermediate-, and low-risk TdP). These categories were based on the risk of drug-associated TdP [21]. We compared the ion channel inhibition against all tested ion currents at 1×, 3×, and 10× free plasma Cmax. Sotalol, which is classified as having a high risk for TdP, inhibited IKr at the clinical free plasma Cmax either exclusively or to a much greater extent than any other currently examined drug. In the intermediate TdP risk category, the extent of blocking IKr by chlorpromazine was, on average, less than that by a drug in the high-risk category, supporting a lower risk of TdP. In the low TdP risk category, mexiletine was associated with greater inhibition of ICa than that of IKr. The extent of APD prolongation was determined from the complex interactions with IKr, ICa, and INa channels, which were differentially altered by tested drugs. This was consistent with the CiPA study, in which high-risk drugs exhibited greater hERG/IKr inhibition than inhibition of other channels, whereas intermediate- and low-risk drugs exhibited greater Cav1.2/ICa inhibition than hERG/IKr inhibition [16]. Our results demonstrated the potential of human iPSC-CMs as valuable tools for predicting drug-induced cardiotoxicities and support their use as part of a tiered testing strategy. Thus, we suggest that human iPSC-CMs could be a comparable or more effective platform than the comprehensive in vitro set of multiple ion channel assays using heterologously expressed channels.
In conclusion, our findings suggest that, compared to heterologous expression systems, human iPSC-CMs efficiently replicated the effects of drugs on cardiac AP and voltage-gated ion channels. Therefore, human iPSC-CMs can replace the established in vitro multiple ion channel assay models and serve as a new human-based model for drug safety pharmacological assessment.

 XML Download
XML Download