

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Ischemia/reperfusion (I/R) injury is inevitable in various clinical settings, such as transplantation and surgeries [1]. The infiltration of inflammatory cells into inflamed sites is critical for I/R-induced organ damage [2-4]. Accumulating evidence proposes that the control of migration in inflammatory cells into inflamed sites could be an effective strategy for the prevention of I/R injury [5-7]. In kidneys, I/R is one of the major causes of acute kidney injury (AKI), of which high mortality and morbidity are serious problems in health and requires the development of effective therapeutics [8].
Ras-related C3 botulinum toxin substrate 1 (Rac1), a Rho family of small GTPase, regulates many cellular functions, including reactive oxygen species (ROS) generation, cell proliferation, apoptosis, and cell migration, which is essential for leukocyte infiltration to the inflamed site [9-13]. Studies have demonstrated that Rac1 is associated with I/R injury in various organs, including kidneys [14,15]. In the kidney, Rac1 inhibition, which starts 3 days after I/R (which may be a time to pass the peak of the injury), significantly reduces kidney fibrosis assessed after 21 days by reducing expressions of profibrotic cytokines and chemokines [16]. Myeloid-selective Rac1 knock-out blocks both cytokine production and macrophage infiltration in angiotensin II-, salt-, and obese-induced kidney injury [17,18]. These indicate that Rac1 is involved in the recovery or fibrosis of damaged kidneys as well as chronic kidney injury by inflammatory responses including macrophage migration. Interestingly, a recent study presents that myeloid-selective Rac1 deletion suppresses lipopolysaccharide (LPS)-induced kidney injury by inhibiting cytokine production, without blocking infiltration of macrophages [19]. These data clearly show that Rac1 plays an important role in inflammatory responses. However, the role of Rac1 on AKI or acute inflammatory cell migration after kidney I/R has not been defined clearly.
Kidneys consist of various, including kidney tubular epithelial cells, endothelial cells, interstitial fibroblasts, and leukocytes. After I/R, the population of cells in kidneys alters; although it is depended on the degree of injury and time after I/R, interstitial cells such as macrophages increase early after I/R, whereas tubular epithelial cells decrease [20]. So, we hypothesize that pretreatment of Rac1 inhibitor protects the kidney against I/R-induced AKI by macrophage infiltration into the kidneys. Here, we investigated the effect of pretreatment of Rac1 inhibitor on kidney I/R injury and monocytes/macrophages migration. Here, we report that Rac1 activity, but not expression, increased in kidneys after I/R, and Rac1 inhibition reduced kidney I/R injury, concomitant with decreased macrophage infiltration.
METHODS
Animal preparation
All animal experiments were conducted under the guidelines provided by the Animal Care and Use Committee of Kyungpook National University (2022-0335). Eight-week-old C57BL/6 male mice (Koatech) were used in experiments. Mice were provided free access to water and standard mouse chow. Animal surgeries were performed as described previously [21]. Before the operation, mice were anesthetized by intraperitoneal injection of pentobarbital sodium (50 mg/kg body weight; ENTOBAR). To induce ischemia, kidneys were exposed via flank incisions, and both renal pedicles were clamped for 25 min using microaneurysm clamps (Roboz Surgical Instruments). The same procedures, except for the clamping of the renal pedicles, were used for sham operation. Body temperature was maintained at 36.5°C–37.5°C throughout all surgical procedures by using a temperature-controlled heating device (FHC). Some mice were administered either NSC23766 (an inhibitor of Rac1, 10 mg/kg body weight; Millipore) or 0.9% NaCl (vehicle) daily beginning 3 days before the operation. Operations were performed 1 h after the last injection of NSC23766. NSC23766 is a small-molecule specific antagonist of Rac1, which inhibits the Rac1 activation by interfering with the binding of guanine nucleotide exchange factors (GEFs) [22,23]. Each experimental animal group consisted of more than 4 mice. Kidneys were either snap-frozen in liquid nitrogen for biochemical analysis or perfusion-fixed in paraformaldehyde-lysine-periodate (PLP; 4% paraformaldehyde, 75 mM L-lysine, and 10 mM sodium periodate; Sigma-Aldrich) for histological studies.
Blood chemistry
Blood was drawn from the retroorbital venous plexus at indicated times in the figures using a heparinized capillary glass tube (Kimble chase). Plasma creatinine (PCr) was measured using a Vitros 250 Chemistry Analyzer (Johnson & Johnson).
Histology
PLP-fixed kidneys were embedded in paraffin and cut into 3-μm-thick sections using a microtome (Leica). Kidney sections were stained with Periodic Acid Schiff stain (PAS) according to the manufacturer's instructions. The sections were observed under a microscope (Leica), and images were captured using i-Solution software (IMT). Kidney damage was scored in a blind manner according to the following criteria as previously described [24,25]. More than 10 fields per kidney section were analyzed.
Western blot analysis
Western blot analysis was performed as described previously [21]. Antibodies were anti-Rac1 (Cat. No. 610650, BD Bioscience) and anti-GAPDH antibodies (Cat. No. NB300-221, Novus, Littleton). Densities of immunoblots were quantified using image analysis software ImageJ (National Institutes of Health).
Rac1-GTP pulldown assay
Rac1 activity was determined by GTP-bound Rac1 using the Rac1-GTP pulldown assay kit according to the manufacturer’s instruction (Cat. No. BK035, Cytoskeleton, Inc.).
Cell culture
Madin-Darby canine kidney (MDCK) cells (ATCC) and RAW264.7 (ATCC) cells, murine macrophages, were cultured on 35 mm diameter six well cell culture plates (SPL Life Sciences) in minimum essential medium (MEM) (Corning) with 5% fetal bovine serum (FBS; MP Biomedicals) and 100 unit/ml streptomycin/penicillin (S/P; Welgene Inc.) at 37°C in a humidified atmosphere containing 5% CO2.
Macrophage migration
Macrophage migration was performed as described previously [26]. RAW264.7 cell migration was assayed using Boyden chamber (Corning Costar) that contains a polycarbonate transwell membrane filter (6.5 mm diameter, 8 μm pore size). The number of 2 × 104 cells was plated on the upper chamber in MEM that contained 5% FBS and 100 unit/ml S/P. After, 100 μM of NSC23766 was treated in the upper chamber for 1 h. The lower chamber was treated with 2 nM of monocyte chemoattractant protein-1 (MCP-1; PeproTech) for 4 h to induce cell migration. Cells were stained with hematoxylin and eosin, and migrated cells that remained on the bottom surface were counted after non-migrated cells were scraped from the upper surface of the membrane with a cotton swab. Pictures were taken using a microscope (Leica).
Phalloidin staining
RAW264.7 cells were cultured on 35 mm diameter 6 well cell culture plates including cover glass. The number of 2 × 104 cells grown on cover glass was treated for 1 h with 100 μM NSC23766. Cells were then treated with 2 nM MCP-1. Four hours after MCP-1 treatment, cells were harvested and then subjected to phalloidin staining for F-actin (phalloidin-TRITC; Sigma-Aldrich) as described previously [26,27]. Briefly, 4% paraformaldehyde (PFA) fixed RAW264.7 cells were reacted with 0.1% Triton-X 100 for 5 min to increase permeability and then reacted with 50 mg/ml phalloidin-TRITC diluted in PBS for 40 min at room temperature. Visualization of nuclei was induced via DAPI staining. Images were observed with a microscope (Leica) and i-Solution software.
RESULTS
Rac1 expression and activity in the kidneys subjected to either sham-operation or I/R
First, we defined the pattern of Rac1 expression in kidneys by immunohistochemical staining using an anti-Rac1 antibody. In sham-operated kidneys, Rac1 was found in the kidney tubular epithelial cells showing cytosol expression (Fig. 1A). Next, we determined Rac1 expression in I/R-injured kidneys. As expected, ischemia caused loss of brush border and nuclei, and detachment of tubular cells from tubule 4 and 24 h after reperfusion, consequently resulting in dilation, thinning, congestion, and cell loss in tubules (Fig. 1B). In addition, I/R gradually increased interstitial cell numbers including F4/80, a marker of monocyte/macrophage, -positive cells over time (Fig. 1C, D). Consistent with kidney histological impairments, I/R increased PCr concentrations (Fig. 1E). In I/R-injured kidneys, Rac1 expression decreased in the tubules in association with a decrease of numbers and damage in tubule cells, while the number of Rac1-positive cells increased in the interstitium in association with an increase in interstitial cell numbers (Fig. 1F).
Moreover, we determined the amount of Rac1 activity and expression in the whole kidney lysates by Rac1-GTP pulldown assay and Western blot analysis, respectively. The expression levels of Rac1-GTP in I/R-injured kidneys were significantly greater than those in sham-operated kidneys (Fig. 1G). However, the level of total Rac1 in I/R-injured kidneys was not significantly changed when compared with that in sham-operated kidneys (Fig. 1G). In addition, we found that most of the F4/80-positive cells expressed strong Rac1-positive signals in the serial sections of the kidneys (asterisks indicate the same tubule, Fig. 1H).
Inhibition of post-I/R kidney damage and macrophage migration into the kidney by Rac1 inhibition
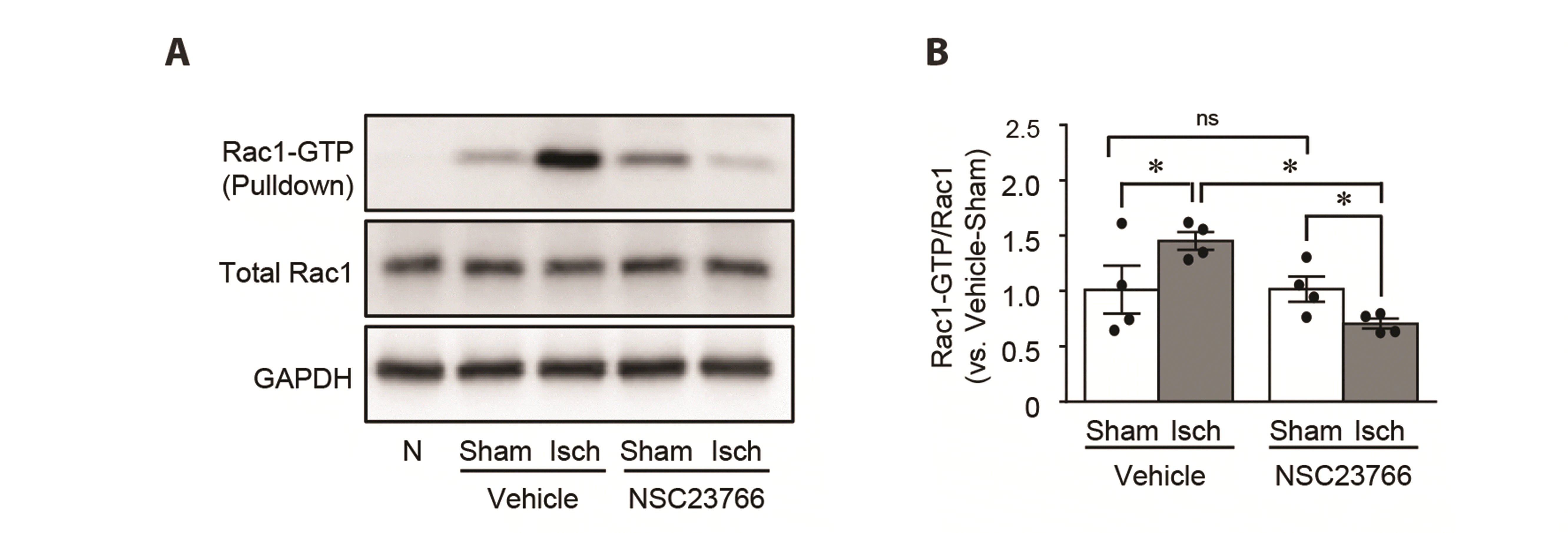
To test whether Rac1 inhibition protects the kidney against I/R injury, we administered either NSC23766, a specific inhibitor of Rac1 activation, or vehicle to mice daily beginning 3 days before the operation. First, we determined the Rac1 activity after the administration of NSC23766. Ischemia significantly increased Rac1-GTP expression 24 h later (Fig. 2). This increase was prevented by NSC23766 pre-administration (Fig. 2). However, I/R in both NSC23766 and vehicle-administered mice did not induce changes in the total Rac1 expression (Fig. 2). NSC administration did not affect Rac1-GTP and total Rac1 expression in sham-operated mice (Fig. 2).
Next, we determined the effect of NSC23766 on the macrophage infiltration into the injured kidney by immunohistochemical staining using anti-F4/80 antibody. I/R increased F4/80-positive cell numbers in the interstitium when compared to sham operation (Fig. 3A, B). Consistent with increases in F4/80-positive cells in the interstitium, I/R increased Rac1-positive cells in the interstitium (Fig. 3A, B). NSC23766 administration inhibited the I/R-induced increases in interstitial Rac1-positive cells and interstitial cells (Fig. 3C, D). NSC23766 administration in sham-operated mice did not induce significant changes in the numbers of Rac1-positive tubule cells and interstitial cells in the kidneys, compared to those in vehicle administration (Fig. 3A-D).
Finally, we evaluated the effect of NSC23766 on kidney I/R-induced histological and functional damage. Kidney I/R-induced tubular cell damage was concomitant with increased PCr concentration (Fig. 3E-G). NSC23766 significantly reduced these tubular cell damage and PCr concentration increase (about 30% in tubular damage; about 30% in PCr) (Fig. 3F, G). NSC23766 administration in sham mice did not induce any significant changes in PCr and kidney morphology (Fig. 3E-G). These results indicate that Rac1 inhibition protects the kidney against I/R insult through regulation of macrophage migration.
Suppression of MCP-1-stimulated RAW264.7 cells migration by Rac1 inhibition
We assessed the effect of Rac1 on the lamellipodia and filopodia formation and migration of RAW264.7 cells. MCP-1 (a chemoattractant cytokine) treatment into the lower chamber activated the migration of RAW264.7 cells across the membrane (Fig. 4A, B). NSC23766 treatment significantly reduced cell migration induced by MCP-1 (Fig. 4A, B). MCP-1 activated the extension of lamellipodia and filopodia (Fig. 4C). These extensions of lamellipodia and filopodia were significantly inhibited by NSC23766 treatment (Fig. 4C). NSC23766 slightly inhibited the formation of lamellipodia in the MCP-1-non-treated cells (Fig. 4C). These data indicate that Rac1 activation stimulates macrophages migration by the activation of lamellipodia and filopodia formation.
DISCUSSION
In this study, we found that ischemia followed by reperfusion activates Rac1 in the kidney, concomitant with increases of interstitial macrophages, and Rac1 inhibition attenuates kidney I/R injury and macrophage infiltration. In addition, Rac1 inhibition suppressed the MCP-1-induced lamellipodia formation and migration of cultured monocytes/macrophages. These results indicate that Rac1 inhibition by pretreatment of Rac1 inhibitor protects the kidney against I/R by the inhibition, at least in part, of macrophage migration, suggesting that macrophage Rac1 could be a therapeutic target for I/R-induced AKI.
Previous studies have shown that kidney I/R activates Rac1 in the kidneys, suggesting that Rac1 inhibition attenuates kidney I/R-induced injury and fibrosis [16,28]. Along the same lines, we found in the present study that 25 min of ischemia increases Rac1-GTP without change of total Rac1 expressions in whole kidney lysates 24 h after reperfusion, and that Rac1 inhibition mitigates kidney I/R damage. Interestingly, in present histological studies, we observed that Rac1 expression appeared to decrease 24 h after I/R in the tubular epithelial cells, whereas its expression in the interstitium increased in a positive correlation with an increase of F4/80-positive macrophages. Although tubule cells expressed low Rac1 expression after I/R, we did not find any significant changes in total Rac1 levels in kidney whole cell lysates. This indicates that unchanged total Rac1 expression levels in the kidneys after I/R injury may be due to increased Rac1-expressing interstitial cell numbers and the difference in Rac1 amount between tubule cells and macrophages. In the present study, we found that most of F4/80-positive cells in the interstitium express Rac1. Also, in the present study, we found that RAW264.7 cells have a relatively much greater amount of Rac1 than MDCK cells, which are kidney tubular epithelial cells (Supplementary Fig. 1). Similarly, Liang et al. [16] reported that 30 min of kidney ischemia increases Rac1-GTP without change of total Rac1 expression in 21 days post-ischemic whole kidney lysates, of which kidneys contain high levels of macrophages compared to normal kidneys [29]. Our results suggest that to define the detailed role of Rac1 in the kidney I/R injury, the role of Rac1 should be defined in both tubule cells and macrophages.
I/R injury is highly associated with the infiltration of circulating leukocytes into inflamed sites. Circulating leukocyte migration into the inflamed sites is regulated by cytoskeleton rearrangements, including formation and extension of lamellipodia and detachment of the adhesion and retraction. Studies have demonstrated that Rac1 is critical for the migration of leukocytes and its inhibition attenuates macrophage migration cells [16,30]. Liang et al. [16] reported that Rac1 inhibition suppresses cytokines and chemokine production and protects the kidney against I/R-induced fibrosis, by inhibiting macrophage accumulation [16,19]. However, several studies have shown that, although Rac1 inhibition affects migration, invasion, and chemotaxis, Rac1 is not essential for the migration or chemotaxis of macrophages [31-34]. However, Nagase et al. [19] reported that Rac-1 deficiency in the mouse myeloid lineage cells reduces LPS-induced kidney injury by reduction of IL-6 and TNF-α production and oxidative stress. In addition, they reported that Rac-1 deficiency in myeloid lineage did not block LPS-induced F4/80-positive macrophage accumulation in the kidney, but Rac1 inhibition in RAW264.7 cells by a pharmacological approach using EHT1864 blocks LPS-driven cytokine induction [19]. However, our data show that Rac1 inhibition by NSC23766 reduced the infiltration of macrophages into the kidneys after I/R and suppressed this MCP-1-induced lamellipodia extension and the migration of macrophages. The extension of lamellipodia is critical for cell movements [35,36]. Therefore, we speculate that this discrepancy between our result and Nagase’s result may be due to experimental settings. As mentioned above, although it is controversial whether the Rac1 of macrophage is essential or not for macrophage infiltration in injured kidneys, our data clearly show that Rac1 inhibition mitigates macrophage migration and lamellipodia formation.
Several studies have suggested that the protective effect of Rac1 is associated with various mechanisms, such as inhibitions of oxidative stress, apoptosis, and NFkB signaling [28,37]. Interestingly, in the present study, we found that Rac1 expression is differently altered between tubules and interstitium; Rac1 expression decreases in the kidney tubules, whereas its expression increased in the interstitium. This suggests that to define the exact role of Rac1 on AKI, our efforts are required in both tubule cells and macrophages, considering that relatively, the role of tubule cell specific-Rac1 is less studied than macrophages-specific Rac1 target studies. However, our data clearly show that monocyte/macrophage Rac1 could be a target for the treatment of I/R-induced AKI.



 XML Download
XML Download