

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Diabetic kidney disease (DKD) is a major complication of diabetes and the most prevalent cause of end-stage renal disease (ESRD). The prevalence of DKD is on the rise, as about 30% of patients with type 1 diabetes mellitus and 40% of patients with type 2 diabetes mellitus develop DKD [1]. In addition, the percentage of patients with ESRD caused by diabetes increased from 22.1% in 2000 to 31.3% in 2015 worldwide [2]. Compared to individuals who only have diabetes, patients with both diabetes and DKD have a mortality risk that is 3- to 12-fold higher, and 90% of DKD patients die before progressing to ESRD [3]. Therefore, it is crucial to unravel the underlying pathogenesis of DKD and develop therapeutic strategies.
Ferroptosis is a recently discovered type of programmed cell death that is linked to the pathogenesis of various diseases [4-6]. Ferroptosis is characterized by iron overload, the accumulation of lethal lipid hydroperoxides, a weakened antioxidant capacity, and mitochondrial changes such as smaller mitochondria, diminished or absent mitochondrial cristae, and ruptured mitochondrial outer membranes [7,8]. These manifestations are distinct from those of apoptosis, necroptosis, and pyroptosis [9]. In ferroptosis, lipid peroxidation is caused by reactive oxygen species (ROS) derived from the Fenton reaction, and the accumulation of these peroxides on cellular membranes can damage the membranes and cause cell death [10]. In addition, lipid peroxidation end-products, such as malondialdehyde (MDA), may form toxic adducts with proteins and DNA, leading to significant cytotoxicity and inducing ferroptosis [11]. Physiologically, lipid hydroperoxides are reduced to hydroxy derivatives by glutathione peroxidase 4 (GPX4) and glutathione (GSH), but during ferroptosis, GSH production is reduced, and GPX4 is unable to exert its normal antioxidant capacity due to the disruption of Xc− transporter [12]. Moreover, ferritin is also involved in the suppression of ferroptosis by storing iron ions [13,14]. Ferritin consists of a heavy chain (FTH) and a light chain (FTL). FTH mediates the conversion of ferrous iron (Fe2+) to the ferric form (Fe3+), which is stored in FTL [15]. Recent evidence points to the role of ferroptosis in the pathology of diverse kidney diseases [16]; specifically, it has been linked to DKD, although the mechanism is not clear [17].
Ferroptosis is regulated by multiple metabolic pathways [18,19]. For example, lipid metabolism pathways can modulate the sensitivity of ferroptosis in various diseases [20,21]. Previous studies have found that a reduction in fatty acid β-oxidation (FAO) increased the susceptibility of renal cell carcinomas to ferroptosis [22]. Typically, mitochondrial FAO consumes most of the fatty acids and reduces lipid peroxidation [23]. However, FAO is reduced in renal carcinomas, increasing their susceptibility to ferroptosis [22]. In patients with diabetes, dyslipidemia is a risk factor for the progression of DKD [24,25]. Abnormal lipid deposition and lipid droplets in glomeruli and tubules were caused by the downregulation of genes for lipolysis and FAO [26,27]. However, it remains unclear whether impaired FAO and lipid accumulation can cause progression to DKD by increasing ferroptosis.
Fatty acid binding protein 4 (FABP4) plays a vital role in lipid metabolism and oxidative stress and contributes to the pathogenesis of a wide variety of metabolic diseases [28,29]. Recent studies have shown that the upregulation of FABP4 promoted the progression of kidney disease [30]. Furthermore, in unilateral ureteral obstruction-induced fibrotic kidneys, FABP4 inhibition by BMS309403 (BMS) rebalanced abnormal lipid metabolism by restoring impaired FAO in tubular epithelial cells and prevented kidney injury [31]. FABP4 transports free fatty acids to the mitochondria and peroxisomes for FAO, to the endoplasmic reticulum for signal transmission and membrane synthesis, to the nucleus for transcriptional regulation of lipid metabolism-associated genes, and to lipid droplets for storage [32]. FABP4 is mainly expressed in adipocytes and macrophages and plays a significant regulatory role in energy metabolism and inflammation [33].
Clinical studies have shown a negative correlation between serum FABP4 levels and the glomerular filtration rate in DKD patients [34], and FABP4 could be used as a useful prognostic marker for adverse renal outcomes in patients with diabetes [35]. In addition, elevated expression of FABP4 in mesangial and renal tubular epithelial cells may be involved in various kidney diseases, such as acute kidney injury, hyperuricemic nephropathy, and DKD [30]. In renal fibrosis, the expression of FABP4 in renal tubules is upregulated, accompanied by the accumulation of lipid droplets and the downregulation of FAO-associated genes, namely peroxisome proliferator activated receptor gamma (PPARG) and carnitine palmitoyltransferase-1 (CPT1) [31]. The FABP4 inhibitor BMS restored impaired FAO, reduced lipid accumulation, and prevented kidney injury [31]. Furthermore, a recent study demonstrated that the expression of FABP4 in the retinal tissue of streptozotocin-induced diabetic mice was dramatically elevated, and the inhibition of FABP4 alleviated lipid peroxidation and oxidative stress in diabetic retinopathy by regulating PPARG-mediated ferroptosis [36]. These results suggest that FABP4 may be involved in regulating ferroptosis, while its specific mechanism remains to be further studied. Herein, our aim is to clarify the role of FABP4 in ferroptosis linked to DKD.
METHODS
Human renal biopsy
Renal biopsies were collected from patients with established DKD at the Department of Nephrology in the Seventh Affiliated Hospital of Sun Yat-sen University. The diagnosis was established with standard histological criteria. Participants with other concomitant renal diseases were excluded. For Prussian blue staining and immunofluorescence staining, renal biopsies samples from patients with glomerular minor lesion served as negative controls (NCs). For transmission electron microscopy (TEM) detection, renal biopsies with the diagnosis of early-stage DKD served as NCs. All biopsy issues were obtained with the consent of the patients and were approved by the Ethics Committee of the Seventh Affiliated Hospital of Sun Yat-sen University (KY-2023-024-01).
Chemicals and materials
Dulbecco’s modified Eagle’s Medium (DMEM)/F12 was obtained from Corning (Corning, NY, USA). DMEM, dimethyl sulfoxide, D-glucose, ferrostatin-1 (Fer-1), and palmitic acid (PA) were acquired from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS) was purchased from Gibco (Waltham, MA, USA). The FABP4 inhibitor BMS was acquired from ApexBio (Houston, TX, USA). Dojindo (Kumamoto, Japan) provided the cell counting kit-8 (CCK-8). Transfection reagents, small interfering RNA (siRNA)-FABP4, si-CPT1A, and its NC were acquired from RiboBio (Guangzhou, China). Oil Red O was obtained from Solarbio Science and Technology (Beijing, China). The fatty acid oxidation assay, DCFDA/H2DCFDA–cellular ROS assay kit, and primary antibodies (anti-GPX4, anti-FTH, and anti-FTL) were acquired from Abcam (Cambridge, UK). Anti-FABP4 and anti-CPT1A antibodies were obtained from Proteintech (Wuhan, China). Anti-AMP-activated protein kinase (AMPK) and anti-phospho-AMP-activated protein kinase (p-AMPK) antibodies were acquired from Cell Signaling Technology (Boston, MA, USA). Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and anti-rabbit/mouse secondary antibodies were acquired from Abclonal (Wuhan, China). Beyotime (Shanghai, China) provided the lipid peroxidation MDA assay kit, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) apoptosis assay kit, and radioimmunoprecipitation assay (RIPA) lysis buffer. Biological assay systems (Hayward, CA, USA) provided the iron assay kit. Nanjing Jiancheng Bioengineering Institute (Nanjing, China) provided the GSH assay kit and superoxide dismutase (SOD) assay kit. The bicinchoninic acid (BCA) protein assay kit and protease inhibitor were obtained from CWBIO (Jiangsu, China). Polyvinylidene fluoride (PVDF) membranes were obtained from Millipore (Billerica, MA, USA). Bovine serum albumin (BSA) and 2.5% glutaraldehyde were purchased from Servicebio (Wuhan, China).
Cell lines
We purchased human renal proximal tubular epithelial (HK2) cells from Cellcook Biotech Co. Ltd. (Guangzhou, China). We cultured the cells with DMEM/F12 supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C in a humidified incubator with 5% CO2. In the control group, cells were cultured with DMEM containing 5 mmol/L D-glucose. To establish high glucose (HG)-induced cells, HK2 cells were exposed to 30 mmol/L D-glucose for 48 hours. In the HG + BMS group, cells were cultured in DMEM supplemented with 30 mmol/L D-glucose and 10 μmol/L BMS for 48 hours. In the HG + Fer-1 group, we cultured cells in DMEM medium containing 30 mmol/L D-glucose and 1 μmol/L Fer-1 for 48 hours.
Cell transfection
RiboBio designed and synthesized the siRNAs targeting FABP4, CPT1A, and the NC. In accordance with the manufacturer’s protocol, cells were transfected with NC-siRNA, FABP4-siRNA, or CPT1A-siRNA at a concentration of 50 nM using transfection reagents from RiboBio. RNA silencing and exogenous expression of FABP4 and CPT1A were optimized with Western blot analysis.
Cell viability assay
First, we exposed HK2 cells to different interventions in 96-well plates with a density of 1×104 cells per well, and cultured them for 24, 48, 72 hours, respectively. Then, each well of cells was incubated with 10 μL of the CCK-8 working reagent for 2 hours at 37°C. The absorbance was detected with a microplate reader (Synergy H1M, BioTek, Winooski, VT, USA) at 450 nm. The change in optical density (OD) relative to the baseline was then calculated by subtracting the average OD of the baseline time point (0 hour) from all OD values.
Prussian blue staining
Human kidney tissue paraffin sections were deparaffinized and rehydrated, followed by staining with Prussian blue solution, and images were captured by an upright optical microscope (Nikon Eclipse E100, Nikon, Tokyo, Japan).
Oil Red O staining
Oil Red O staining was used to detect the deposition of neutral lipids in HK2 cells. Cells were washed with phosphate buffered saline (PBS) twice and fixed in 4% paraformaldehyde at room temperature for 30 minutes. Then, cells were incubated with Oil Red O working solution for 15 minutes in the dark at room temperature. After rinsing with double distilled water (ddH2O) three times, the cells were then stained with hematoxylin for 1 minute and gently washed with ddH2O. Finally, light microscopy was used to visualize the cells, and the images were captured at × 400 magnification.
ROS assay
The production of intracellular ROS levels was detected with a DCFDA/H2DCFDA–cellular ROS assay kit. After treatment with HG, with and without BMS or Fer-1, cells were incubated with 10 µmol/L DCFDA for 30 minutes at 37°C in the dark and visualized with fluorescence microscopy.
Iron content detection
The cells were washed with ddH2O three times and collected with a cell scraper. An ultrasonic crusher was used for homogenization. Supernatants were then collected after centrifuging the cells at 12,000 rpm for 10 minutes at 4°C. We mixed 20 volume units of reagent A, 1 volume unit of ddH2O, and 1 volume unit of reagent C to prepare the working solution. A total of 50 μL of the diluted standards and 50 μL of the sample were transferred into a 96-well plate, and the working solution (200 μL) was added to each well, followed by incubation for 40 minutes at room temperature in the dark. OD values were then immediately measured at 590 nm.
Measurement of MDA content
MDA levels were measured using the MDA assay kit, following the manufacturer’s instructions. Briefly, cells were lysed with RIPA lysis buffer, and the cell lysates were centrifuged at 12,000 rpm for 10 minutes at 4°C to obtain the supernatant. The supernatant of cell lysates (100 μL) was mixed with MDA working solution (200 μL) and incubated at 100°C for 15 minutes. The mixture was centrifuged at 1,000×g for 10 minutes after cooling to room temperature, and supernatant (200 μL) was added to a 96-well plate to measure absorbance at 532 nm.
GSH level evaluation
We collected HK2 cells and used an ultrasonic crusher to obtain a homogenate, which was centrifuged at 12,000 rpm for 10 minutes at 4°C, and the supernatant was collected. Samples and standard (100 μL) were respectively added into a 96-well plate, followed by adding 125 μL of the GSH assay mixture into each well and incubating for 5 minutes at room temperature. The absorbance was detected at the wavelength of 405 nm.
SOD assay
The activity of SOD in HK2 cells was examined with the total SOD assay kit (hydroxylamine method). This assay used the xanthine-xanthine oxidase system to produce superoxide ions, which reacted with hydroxylamine to form nitrite, and appeared purple with a chromogenic agent. The absorbance at 550 nm was determined.
FAO enzyme measurements
The analysis of FAO was performed using the fatty acid oxidation assay kit, which detects the expression of FAO enzymes including long chain 3-hydroxyl-coenzyme A (CoA) dehydrogenase (HADHA), acyl-CoA dehydrogenase very long chain (ACADVL), and acyl-CoA dehydrogenase, medium chain specific (ACADM). Cells were washed with PBS, fixed with 4% paraformaldehyde for 15 minutes, permeabilized with 0.1% Triton X-100 in PBS for 15 minutes, and blocked with 5% BSA for 30 minutes. Primary antibodies (HADHA, ACADVL, ACADM) were incubated overnight at 4°C. A fluorescent secondary antibody was incubated for 1 hour at room temperature. 4′,6-Diamidino-2-phenylindole (DAPI) was then used to stain the nuclei for 5 minutes. Finally, an inverted fluorescence microscope was used to capture images.
Western blotting
Cells were washed three times with pre-chilled PBS, homogenized with RIPA lysis buffer containing a protease inhibitor, and centrifuged at 12,000 rpm for 10 minutes at 4°C to collect the supernatant. 5X loading buffer was added to the protein samples and boiled for 6 minutes at 98°C. After electrophoresis in 10% sodium dodecyl sulfate-polyacrylamide gel, the proteins and markers were transferred onto PVDF membranes. Then, a blocking solution of 5% skim milk was applied to membranes for 1 hour at room temperature, followed by incubation with primary antibodies against FABP4, GPX4, FTH, FTL, AMPK, p-AMPK, CPT1A, and GAPDH overnight at 4°C. After washing three times with TBST, the blots were incubated at room temperature for 1 hour with the secondary antibody. Finally, immunoblots were visualized using a chemiluminescence reagent, and AlphaEaseFC software (Alpha Innotech, San Leandro, CA, USA) was used for quantitative densitometry of the protein bands.
Immunofluorescence assay
Immunofluorescence staining was used to detect the expression of FABP4, GPX4, FTH, FTL, and CPT1A in human kidney tissues. The tissue slices were incubated overnight at 4°C with anti-FABP4, anti-GPX4, anti-FTH, anti-FTL, and anti-CPT1A antibodies, respectively. Then, the slices were incubated at room temperature for 50 minutes with the secondary antibody, which was labeled with fluorescein. DAPI was then used to stain nuclei for 10 minutes. Finally, an ortho-fluorescence microscope (Nikon Eclipse C1) was used to capture images.
Transmission electron microscopy
We fixed HK2 cells with 2.5% glutaraldehyde for 2 to 4 hours at 4°C and wrapped them with 1% agarose. Next, cells were fixed with 1% aqueous osmium tetroxide for 2 hours, dehydrated in gradual ethanol (50% to 100%) and 100% acetone, followed by embedding with embedding agents in an oven at 37°C overnight and curing at 60°C for 48 hours. We cut ultrathin sections of 60 to 80 nm and stained them with 2% uranyl acetatesaturated alcohol solution and 2.6% lead citrate for 15 minutes. The sections were dried overnight at room temperature. Eventually, we used TEM (HT7700-SS, Hitachi, Tokyo, Japan) to observe and capture images.
Statistical analysis
Prism version 8.0 (GraphPad, La Jolla, CA, USA) was used to perform statistical analyses. The results were presented as mean±standard deviation. One-way analysis of variance followed by the Tukey multiple-comparison test was applied for multiple comparisons. The Student’s t test was used for comparisons between two groups. P values <0.05 were considered to indicate statistical significance.
RESULTS
DKD patient biopsies
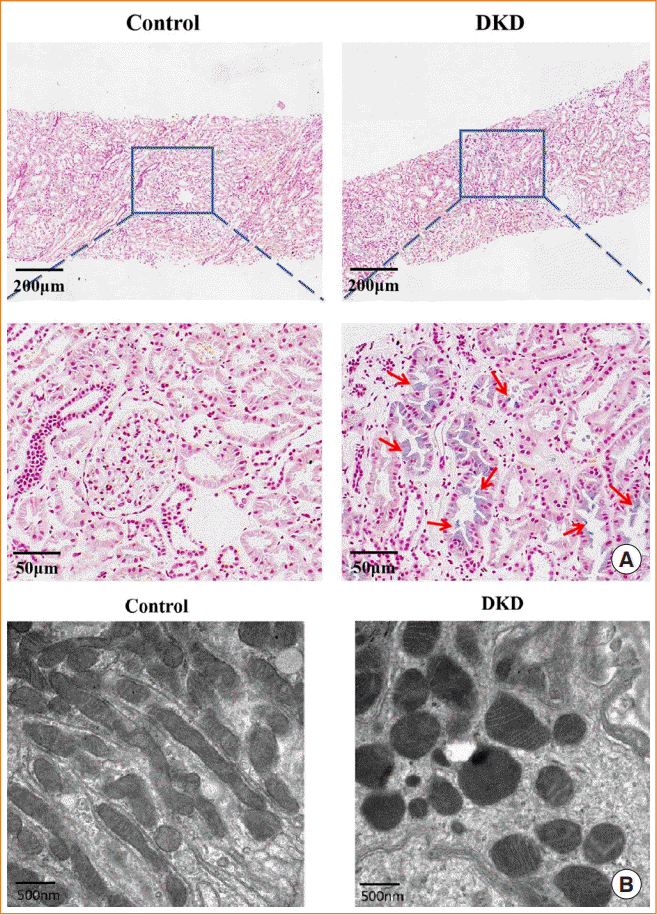
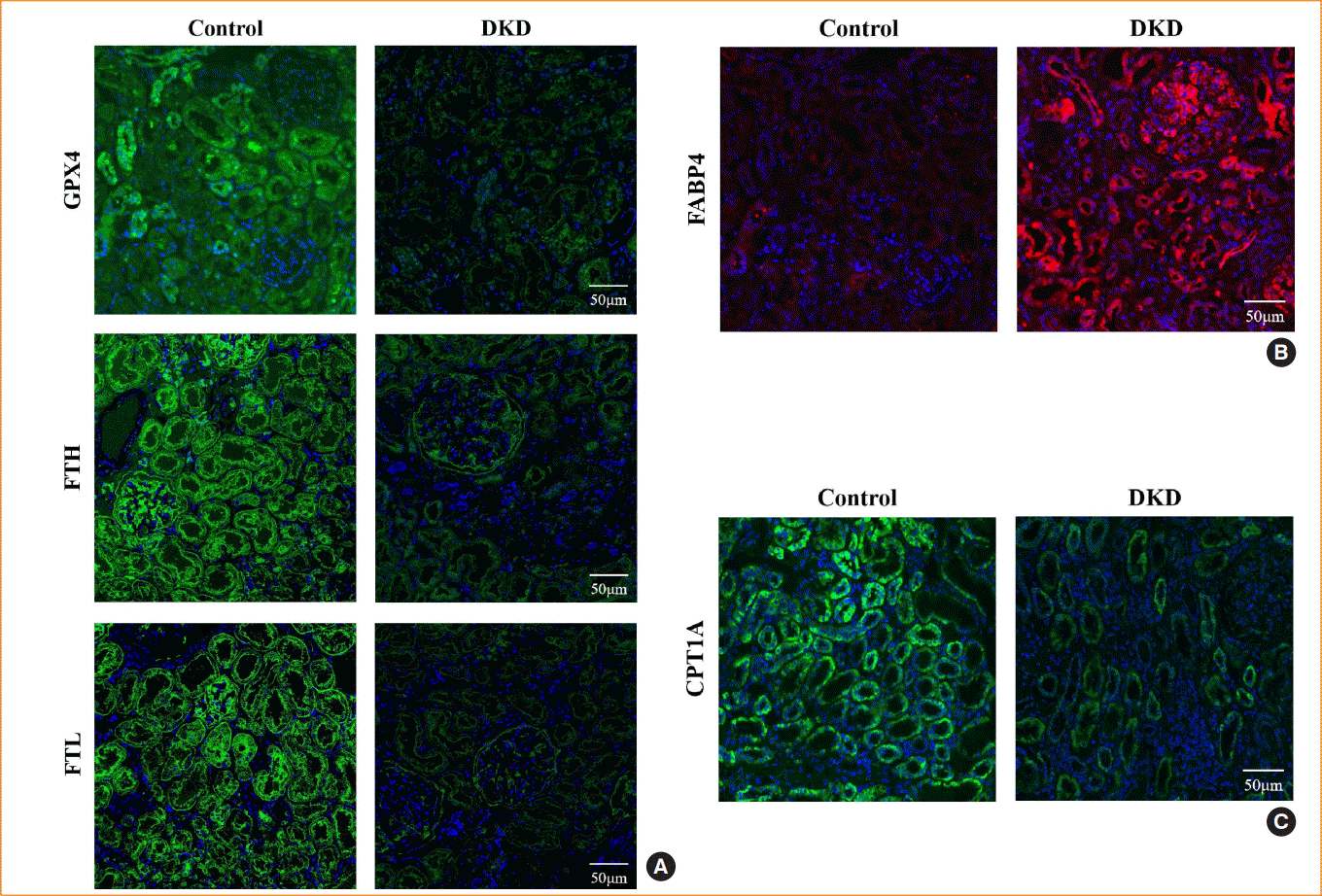
In DKD patients, the renal tubules showed iron accumulation, as detected by Prussian blue staining (Fig. 1A), whereas TEM showed reduced mitochondrial volume and loss of cristae (Fig. 1B). These morphological changes are consistent with ferroptosis. In addition, the expression of ferroptosis-related proteins, including GPX4, FTH, and FTL, were reduced (Fig. 2A). These alterations in ferroptosis-related indicators suggest that ferroptosis is involved in diabetic kidney injury. In kidney biopsy tissues, the expression of FABP4 was upregulated in both renal tubules and glomerulus, compared to non-DKD patients (Fig. 2B), whereas the expression of CPT1A was reduced (Fig. 2C).
Involvement of ferroptosis in HG-induced HK2 cells
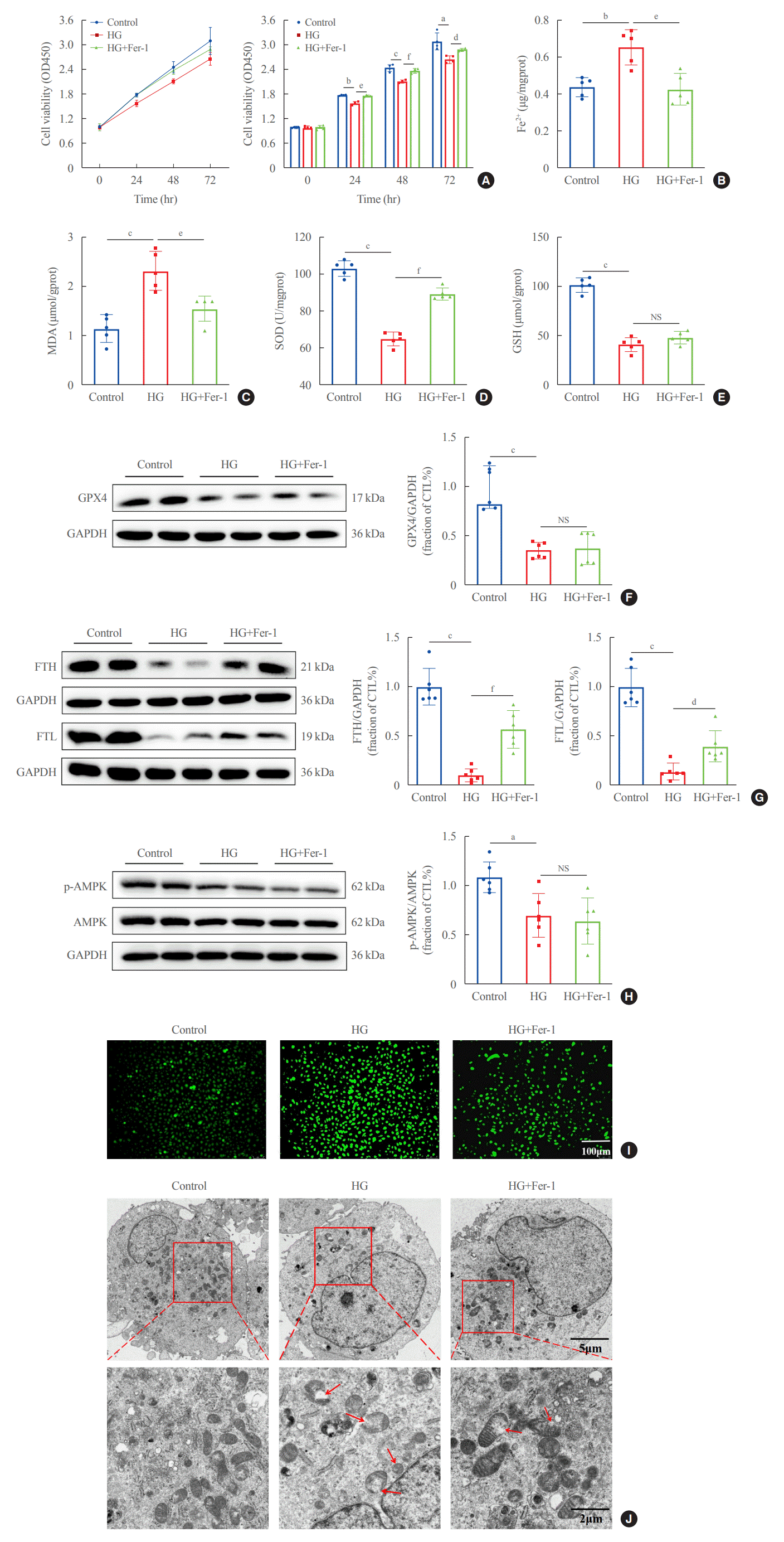
To explore whether ferroptosis is involved in HG-induced renal tubule injury, we treated HK2 cells with the ferroptosis inhibitor Fer-1 in medium containing HG for 48 hours. HG-HK2 cells were less viable than the control group, and exposure to Fer-1 increased the cell viability (Fig. 3A). In the HG group, the levels of Fe2+ and the lipid peroxidation product MDA were increased, whereas SOD and GSH levels and the expression of GPX4, FTH, FTL, and p-AMPK were decreased (Fig. 3B-H). HK2 cells were stained with DCFDA/H2DCFDA to detect ROS generation. Compared to the control group, intracellular ROS levels were increased in HG-HK2 cells, but decreased after Fer-1 treatment (Fig. 3I). HG-treated cells showed mitochondrial cristae reduction or even disappearance, and mitochondrial outer membrane rupture (Fig. 3J). In summary, ferroptosis inhibitor Fer-1 reverted the changes induced by HG, suggesting that HG could induce ferroptosis in HK2 cells.
Involvement of FABP4 in HG-induced ferroptosis in HK2 cells
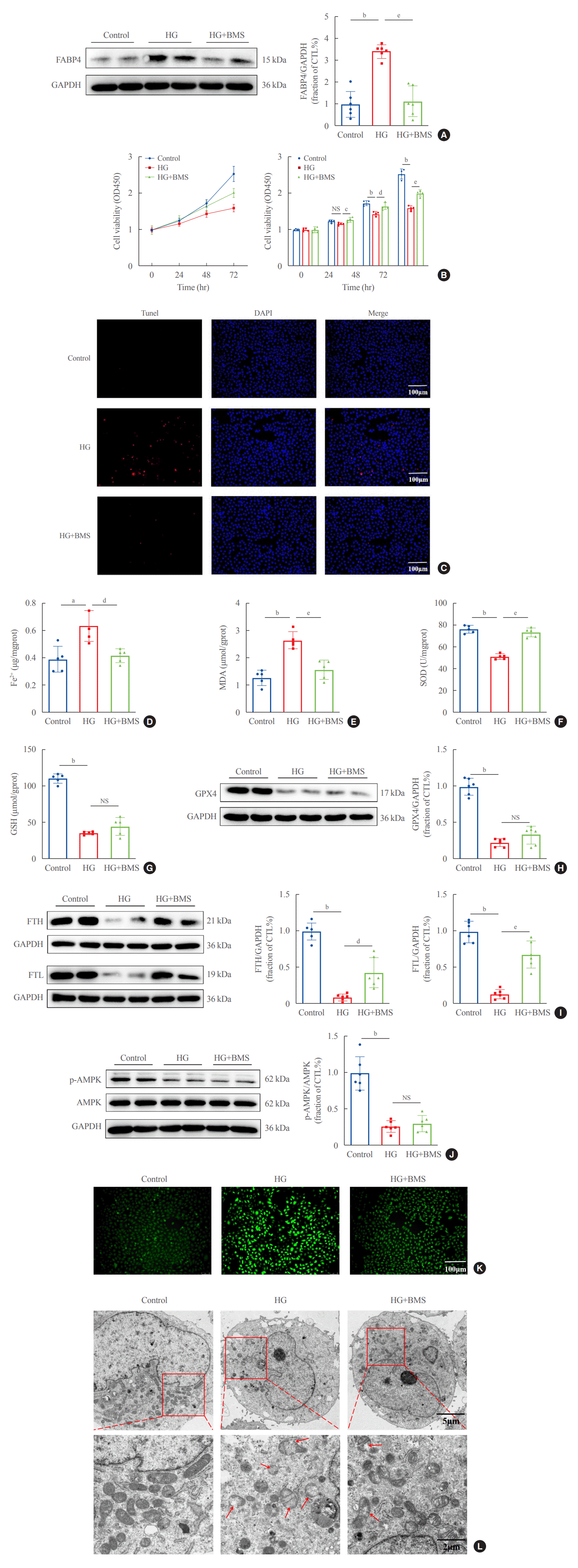
To explore whether FABP4 is involved in HG-induced ferroptosis, HK2 cells were treated with the FABP4 inhibitor BMS. FABP4 expression was clearly upregulated in HG-HK2 cells and decreased after BMS treatment (Fig. 4A). FABP4 inhibition reverted the changes induced by HG: increased cell viability (Fig. 4B), reduced cell death (Fig. 4C), reduced abnormally high intracellular levels of Fe2+ (Fig. 4D), MDA (Fig. 4E), and ROS (Fig. 4K), increased abnormally low levels of SOD (Fig. 4F), increased FTH and FTL expression (Fig. 4I), and attenuated morphological changes in mitochondria (Fig. 4L). However, there was no statistically significant difference in changes of GSH and GPX4 after BMS treatment (Fig. 4G, H). Thus, FABP4 inhibition reversed the characteristic effects of ferroptosis in HG-HK2 cells. In addition, we also measured p-AMPK levels in HK2 cells under HG conditions. The results indicated that the expression of p-AMPK decreased in HG-HK2 cells. However, BMS could not reverse this change (Fig. 4J).
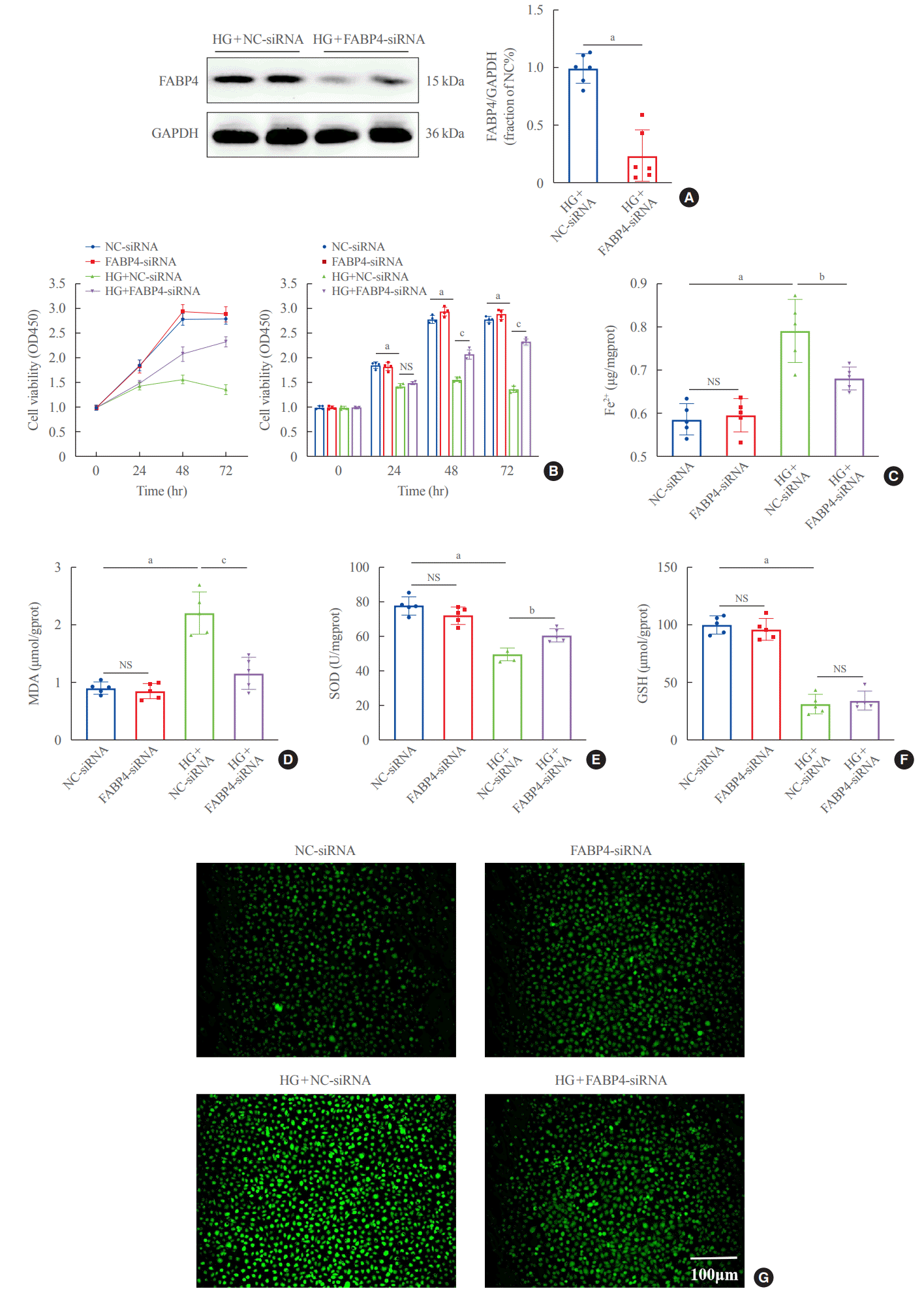
To further clarify the effect of FABP4 on ferroptosis, HG-HK2 cells were transfected with FABP4-siRNA. Western blots showed that FABP4 was downregulated after transfection (Fig. 5A). Similarly, FABP4 silence significantly increased the cell viability under HG conditions (Fig. 5B). Besides, lower levels of Fe2+, ROS and MDA, and increased levels of SOD were also detected in the HG + FABP4-siRNA group compared with the HG + NC-siRNA group (Fig. 5C-G). The above results suggested that FABP4 was involved in HG- induced ferroptosis in HK2 cells and inhibition of FABP4 could protect cells from ferroptosis.
Mediation of HG-induced ferroptosis by FABP4, most likely through inhibiting FAO
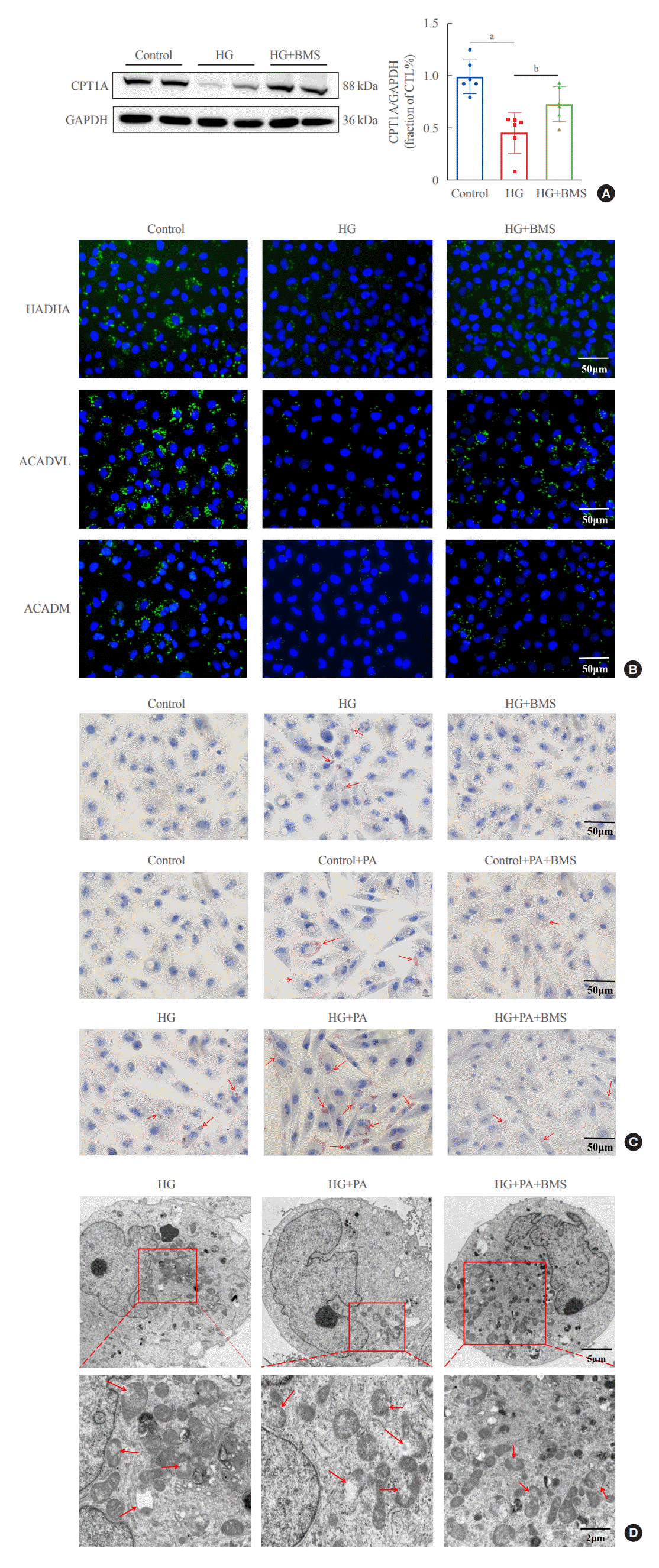
Since the inhibition of FAO increases the sensitivity to ferroptosis, we explored whether FABP4 regulated HG-induced ferroptosis by inhibiting FAO. In HG-HK2 cells, expression of CP-T1A, the rate-limiting enzyme for FAO, was reduced, and recovered after FABP4 inhibition by BMS (Fig. 6A). As showed by immunofluorescence assays, compared with the control group, the expression levels of key enzymes involved in FAO pathway(HADHA, ACADVL, and ACADM) decreased in HG-HK2 cells, which could be reversed by BMS (Fig. 6B). In addition, the abnormal accumulation of lipid droplets is considered to be a marker for dysregulation of fatty acid metabolism. We also detected the intracellular lipid deposition by Oil Red O staining, which demonstrated that lipid deposition was elevated in HG-HK2 cells. However, this was reduced after FABP4 inhibition by BMS (Fig. 6C). These results collectively indicated that the inhibition of FABP4 could restore impaired FAO induced by HG.
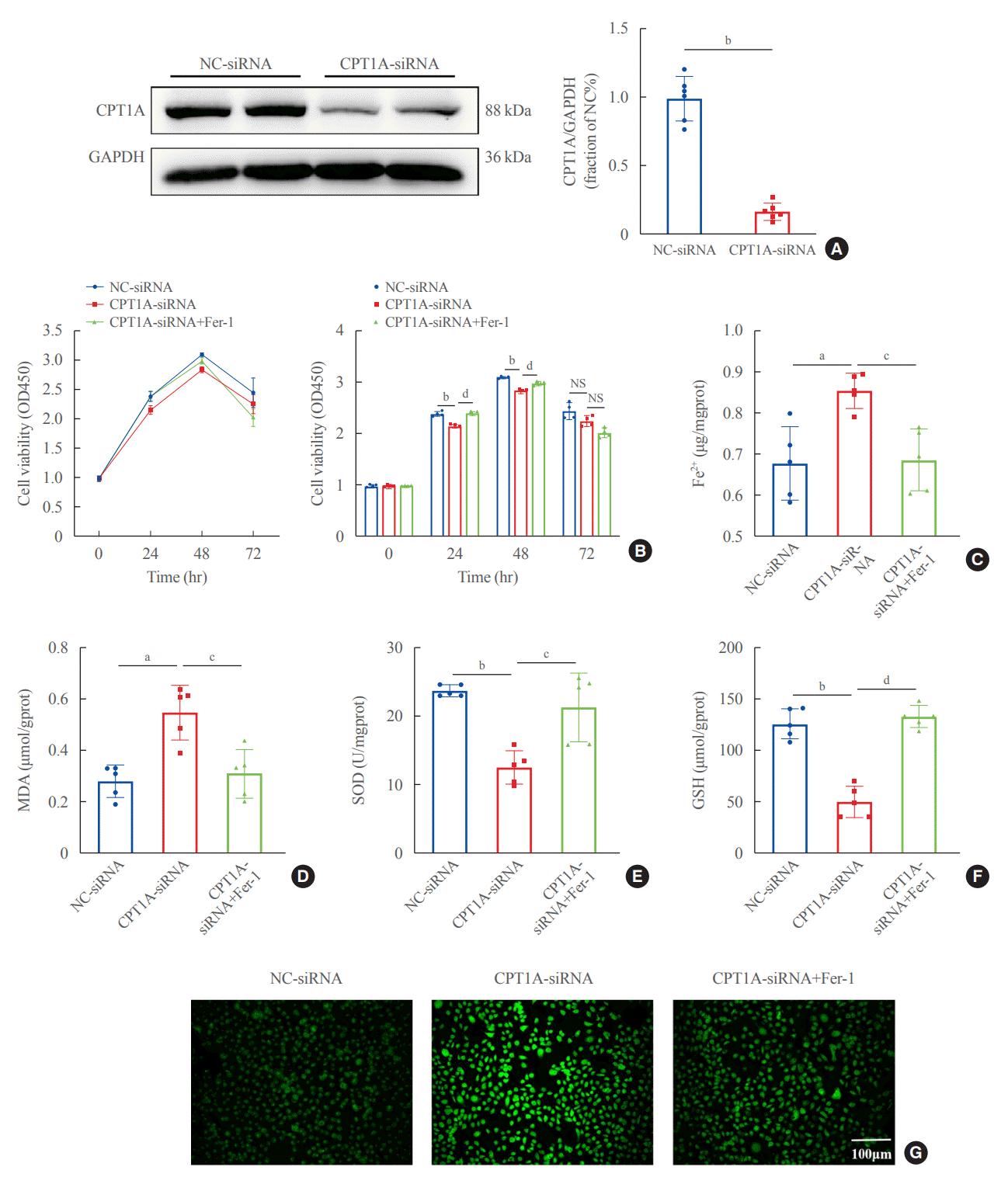
To further characterize the involvement of lipid deposition induced by FABP4 and impaired FAO in HG-induced ferroptosis, we cultured HK2 cells with PA, and treated them with or without BMS. When HK2 cells were exposed to PA, lipid accumulation, and ferroptosis-associated mitochondrial damage were observed, and these effects were also reduced after FABP4 inhibition by BMS (Fig. 6C, D). Furthermore, in order to identify whether the inhibition of FAO could promote ferroptosis in HK2 cells, we also transfected HK2 cells with CPT1A-siRNA and detected ferroptosis-related indicators. Western blot showed that CPT1A was downregulated after transfection (Fig. 7A). After being transfected with CPT1A-siRNA, cell viability significantly decreased (Fig. 7B), the levels of ROS, Fe2+, and MDA increased, whereas SOD and GSH decreased (Fig. 7C-G). Similarly, these changes could be reversed by Fer-1, indicating that inhibition of CPT1A could induce ferroptosis in HK2 cells. Overall, these results suggest a link between impaired FAO induced by FABP4 and ferroptosis in HG-HK2 cells. FABP4 increased the sensitivity of HK2 cells to ferroptosis by inhibiting FAO and aggravating lipid accumulation.
DISCUSSION
In the present study, we showed that the expression of FABP4 was elevated in DKD patients, whereas that of CPT1A, GPX4, FTH, and FTL was reduced. This was accompanied by iron deposition in renal tubules and the disappearance of mitochondrial cristae, characteristic of ferroptosis. Using HG-HK2 cells, we also showed that ferroptosis was involved in HG-induced tubular injury. In HG-HK2 cells, FABP4 was upregulated, whereas FABP4 inhibition abrogated lipid accumulation and peroxidation by restoring FAO. The inhibition of FABP4 increased the resistance to ferroptosis in HK2 cells, suggesting that FABP4 played a regulatory role in the HG-induced ferroptosis of renal tubular cells, and FABP4 may serve as a new therapeutic target in DKD.
Our study showed that the pharmacological and genetic inhibition of FABP4 improved cell viability, prevented ROS and MDA production, and reduced intracellular iron levels in HG-HK2 cells, indicating that inhibiting FABP4 can suppress lipid peroxidation and ferroptosis. FABP4 can mediate ferroptosis and exacerbate the progression of DKD. Indeed, previous reports have shown that FABP4 knockout inhibits inflammation and apoptosis in kidney tubules and improves renal dysfunction in rhabdomyolysis-induced AKI [37]. FABP4 was also found to be involved in ferroptosis in cancer and diabetic retinopathy by regulating lipid metabolism [36,38]. Ferroptosis is regulated by multiple metabolic pathways [39-41], and one main characteristic is lipid peroxidation, which could be driven by abnormal lipid metabolism. It is known that lipid metabolism modulates ferroptosis sensitivity or tolerance in different disease models [42]. Polyunsaturated fatty acids (PUFAs) are most susceptible to lipid peroxidation, and the peroxidation of PUFA-containing membrane phospholipids (PUFA-PLs) is the key trigger of ferroptosis [43]. PUFAs are converted to PUFA-CoA and incorporated into phospholipids to form PUFA-PLs, which are peroxidized by iron-dependent ROS, triggering ferroptosis [42]. The inhibition of FAO increases sensitivity to ferroptosis, probably because it leads to the accumulation of free fatty acids, such as PUFAs, which serve as a substrate for peroxidation [23].
In contrast to PUFAs, monounsaturated fatty acids (MUFAs) can protect cells against ferroptosis by inhibiting the accumulation of lipid ROS in cell membranes and reducing PUFA incorporation into phospholipids [44]. Previous studies have shown that under glucose deprivation conditions, the depletion of adenosine triphosphate (ATP) activates the AMPK pathway, which suppresses the synthesis of MUFAs by phosphorylating and inhibiting acetyl-CoA carboxylase, and thus prevents ferroptosis [45]. Studies have also indicated that AMPK dysregulation plays an essential role in the progression of DKD [46]. Physiologically, when AMPK is activated, it promotes energy-producing catabolic pathways and inhibits anabolic pathways, maintaining the energy metabolism balance [47]. However, in diabetic conditions, the phosphorylation of AMPK is suppressed, which results in abnormal glucose and lipid metabolism [46]. Considering that AMPK activation is responsible for ferroptosis suppression, we also measured the expression level of p-AMPK in HK2 cells under HG conditions. Our results indicated that p-AMPK expression was decreased in HG-HK2 cells, indicating that AMPK phosphorylation was inhibited in HG conditions. However, BMS could not reverse this change, indicating that BMS inhibited ferroptosis via an AMPK-independent pathway.
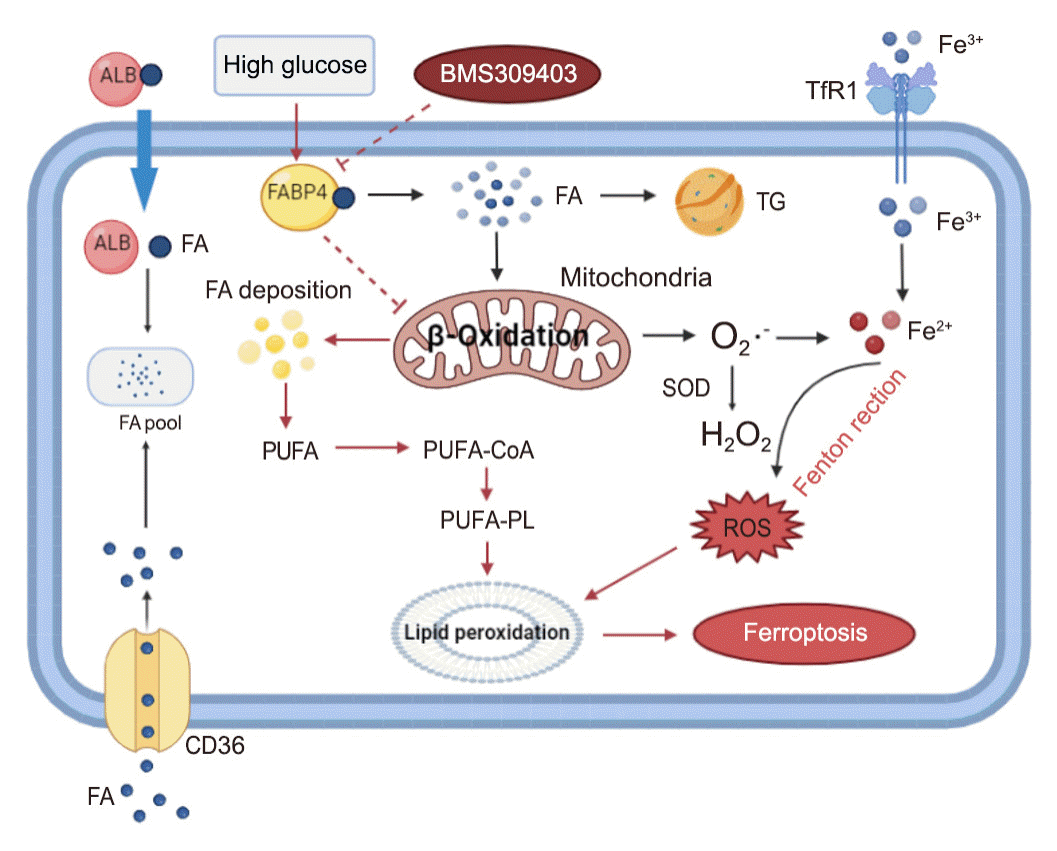
The normal physiological activity of kidney tubule cells requires FAO to generate ATP [48]. Fatty acids are primarily taken up by renal tubules from the basolateral membrane through cluster of differentiation 36 (CD36) receptors and fatty acid transporting protein (Fig. 8), and also through the apical membrane by endocytosis in albumin-bound form [49]. Free fatty acids that enter into the cell are converted to fatty acyl-CoA and transported to mitochondria via CPT1, entering the FAO path-way [50]. Impaired FAO is a primary mechanism of tubular damage and fibrosis in renal disease, and renal tubule-specific overexpression of CPT1 was shown to reduce kidney injury and fibrosis [51]. Herein, we found that HG-HK2 cells exhibited downregulated CPT1A expression and abnormal lipid accumulation, consistent with FAO inhibition. Conversely, FABP4 inhibition restored FAO and attenuated abnormal lipid deposition. We confirmed that the inhibition of FAO by transfecting HK2 cells with CPT1A-siRNA could promote ferroptosis. In addition, by exposing HK2 cells to PA, we also found that lipid deposition could induce ferroptosis. Thus, the inhibition of FABP4 suppresses lipid peroxidation and ferroptosis, partly by restoring FAO and attenuating abnormal lipid accumulation.
In DKD patients, previous studies have shown heavy lipid deposition in podocytes, tubular epithelial cells and mesangial cells, together with the dysregulation of lipid metabolism genes [26]. The dysregulation of the formation, oxidation, and uptake of lipids leads to ectopic lipid accumulation in the kidneys and may contribute to DKD progression [52-54]. Renal lipid deposition may cause renal injury by activating the inflammatory response, ROS production, mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis [24,49]. Herein, we showed that FABP4 triggered ferroptosis by inhibiting FAO, revealing an association between abnormal lipid metabolism and ferroptosis in DKD. Since ferroptosis is triggered by the peroxidation of PUFAs in cell membrane phospholipids [42,43], the accumulation of PUFAs due to impaired FAO could increase lipid peroxidation and thereby induce ferroptosis [22]. Nevertheless, the specific mechanism through which FABP4 mediates ferroptosis by inhibiting FAO remains to be further studied in the future.
In conclusion, our study indicated that FABP4 mediated ferroptosis in HG-HK2 cells by inhibiting FAO. This suggests that targeting FABP4 and ferroptosis in DKD may be a novel therapeutic strategy.
 XML Download
XML Download