

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) are clonal disorders of hematopoietic stem cells; these include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). PV and ET are the most common subtypes of MPNs. In the United States, the incidences of PV and ET are comparable at 1.0–2.0/100,000 person-yr. However, PMF remains uncommon, having an incidence of 0.3/100,000 person-yr [1]. The prevalence of PV and ET is estimated to be 44–57 and 38–57/100,000 people, respectively, in the United States (US) [2]. Caucasians showed a higher incidence of MPNs; East Asians and Africans showed a higher incidence of ET; Caucasians and Hispanics showed a higher incidence of PV [3].
MPNs are inflammatory cancers, wherein the malignant clone generates cytokines that sustain the inflammatory drive in a self-perpetuating vicious cycle. MPNs are linked to organomegaly, cytopenia, and a variety of constitutional complaints, which may significantly impair an individual’s quality of life. The course of MPNs follows a biological continuum, that is, from early cancer stages (ET/PV) to advanced myelofibrosis as well as impending leukemic transformation. Therefore, monitoring the transformation of ET/PV to MF/AML is paramount. However, there are no approved therapeutic regimens to prevent this progression. Additionally, thrombosis, which occurs in 20–30% of the patients, is a major contributor of ET- and PV-associated morbidity and mortality [4]. Therefore, classifying risk categories and attaining suitable thrombosis prevention for each category are the basis of current ET and PV treatments.
CURRENT TREATMENT STRATEGIES FOR PATIENTS WITH PV/ET
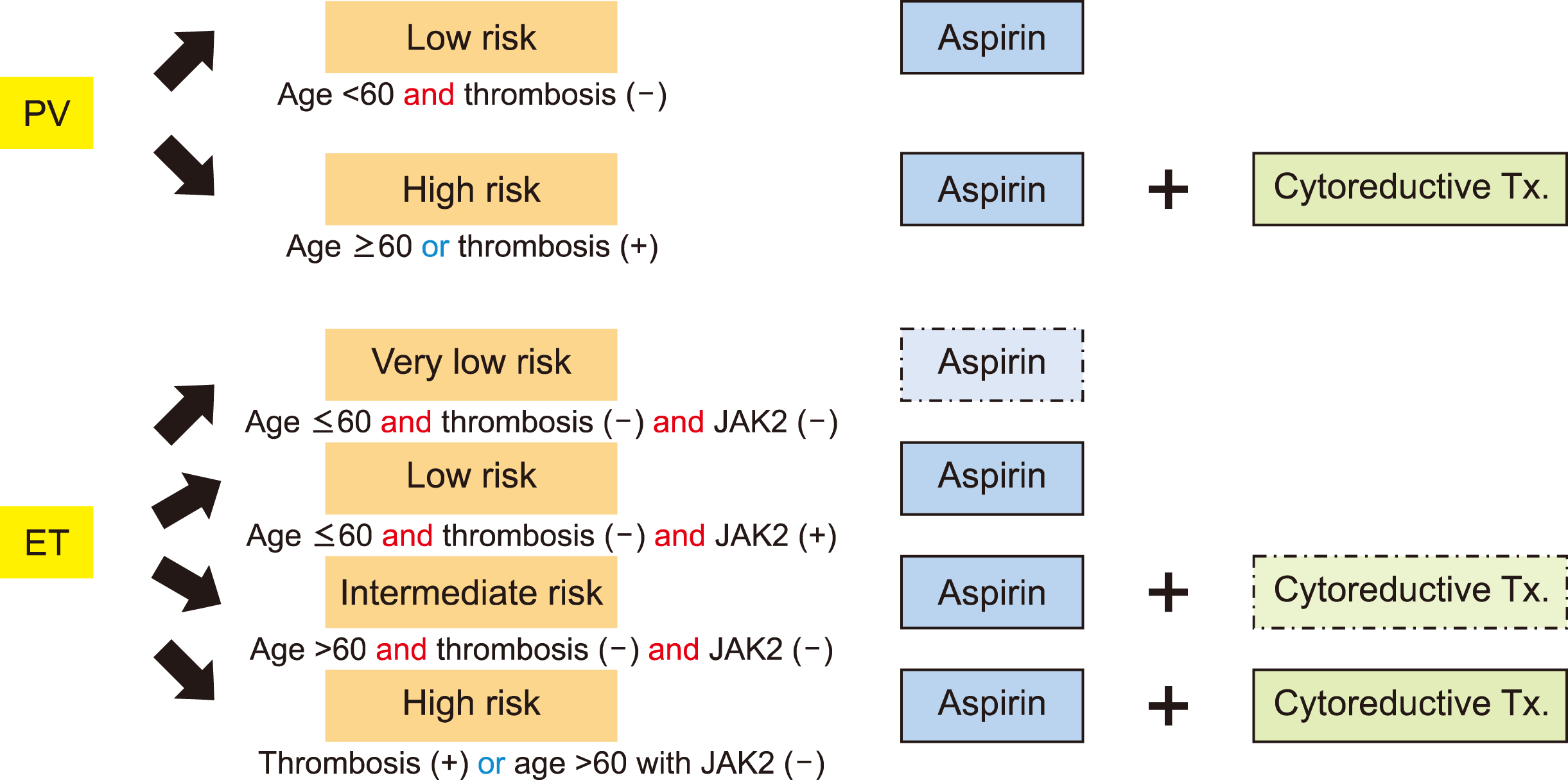
Patients with ET and PV were classified as either “low-risk” or “high-risk” based on their risk for thrombosis. The risk groups defined in the European Collaboration on Low Dose Aspirin in PV (ECLAP) study have now been adopted in other expert consensus guidelines; low-risk PV was defined as those aged ≤60 and with no history of thrombosis, whereas high-risk PV was defined as those aged >60 or with any history of thrombosis [5, 6]. Similarly, the revised International Prognostic Score of Thrombosis for ET (R-IPSET) has been suggested for stratifying ET risk based on expert consensus guidelines [7]. This scoring system categorizes patients into four risk groups: (1) very low risk (age ≤60 yr, no history of thrombosis, and absence of JAK2 V617F mutation); (2) low risk (age ≤60 yr, no history of thrombosis, and presence of JAK2 V617F mutation); (3) intermediate risk (age >60 yr, no history of thrombosis, and absence of JAK2 V617F mutation); and (4) high risk (age >60 yr, presence of JAK2 V617, or any history of thrombosis) (Fig. 1).
In general, low-dose aspirin (81–100 mg/day) is recommended for all patients with PV or ET having no absolute contraindications to bleeding. However, very low-risk patients with ET may not require any treatment unless presenting with cardiovascular risk factors; in this case, once-daily low-dose aspirin therapy is recommended. Moreover, cytoreductive treatment is generally not recommended for low-risk groups, but rather for high-risk groups. However, in certain cases ropeginterferon may be considered, as its efficacy in low-risk PV has been documented [8]. Lastly, intermediate-risk patients with ET may not require cytoreductive treatment unless they have cardiovascular risk factors (Fig. 1) [9].
There is a growing interest in hematologic cancer mutations. These have been exploited in numerous ways for diagnostic, therapeutic, and prognostic purposes. In MPNs, calreticulin (CALR), myeloproliferative leukemia (MPL), and JAK2 mutations are recognized as driver mutations; genetic alterations are known to affect disease progression [10]. The Mutation- Enhanced International Prognostic Scoring Systems (MIPSS) for PV and ET were established by analyzing 906 patients that were molecularly annotated (404 PV and 502 ET) from Mayo Clinic (N=416) and the University of Florence (N=490). In a multivariate analysis, the following characteristics negatively impacted survival: (1) in PV, age >67 yr (2 points), SRSF2 mutation (3 points), thrombosis history (1 point), and leukocyte count ≥15×109/L (1 point); and (2) in ET, age >60 yr (4 points), male sex (1 point), leukocyte count ≥11×109/L (1 point), and SRSF2/SF3B1/U2AF1/TP53 mutations (2 points). The resultant three-tiered MIPSS-PV and MIPSS-ET models stratified patients into the following groups: (1) low (PV: 0–1 points, ET: 0–1 points; median survival not reached in PV and 34.4 yr in ET); (2) intermediate (PV: 2–3 points, ET: 2–5 points; median survival: 10.3 yr in PV and 14.1 yr in ET); and (3) high-risk (PV: 4 points, ET: 6 points; median survival 4.6 yr in PV and 8.3 yr in ET) [11]. This was added to the consensus guidelines of the National Comprehensive Cancer Network (NCCN) for 2022 [6].
In addition to age, history of thrombosis, and mutation, other evaluation tools have also been investigated for classifying and prognosticating ET and PV risk groups. In particular, numerous studies have shown that leukocytosis affects the prognosis of patients with PV and ET. However, these findings remain ambiguous [12-14]. Recent literature indicated that persistent leukocytosis may predict disease progression in patients with PV [15]. This trend was observed during the follow-up period as opposed to a single test, thus providing a meaningful benefit. Furthermore, several studies on the neutrophil-to-lymphocyte ratio (NLR) in patients with MPNs have also been published. NLR, which is the ratio of the absolute neutrophil and absolute lymphocyte counts, is a rapid and simple method for assessing inflammatory status and has the potential for predicting inflammation and mortality in a variety of diseases [16]. As mentioned previously, MPNs are inflammatory cancers in which a malignant clone triggers the production of inflammatory cytokines. In this regard, an increase in NLR was predicted in patients with MPNs, as shown in several studies [17, 18]. Some studies have reported poor prognosis in the group with a higher NLR in patients with MPN [17, 19]. Recently, Carobbio et al. [20] investigated NLR as a predictor of thrombosis in PV. They showed that the risk of venous thrombosis was independently associated with a history of previous events (HR=5.48, P≤0.001) and an NLR ≥5 (HR=2.13, P=0.001). In addition, the relative risk in both the low- and high-risk groups almost doubled in the presence of NLR ≥5. These results were confirmed in two separate external cohorts of patients with PV from Italy (Florentine, N=282; Rome, N=175). Furthermore, there were numerous incidences of thrombosis among low-risk PV patients with a high NLR, thereby indicating that the present risk category had limitations. Studies have also shown similar results in the validation cohort, thus also demonstrating its meaningful benefit. Some studies have investigated the relationship between NLR and PV diagnosis [21]. Among 240 patients with erythrocytosis who were tested for JAK2 mutation, 70 had PV and 170 had secondary polycythemia. The median NLR was significantly higher in the PV group than in the secondary polycythemia group (6.04 vs 1.77, P<0.001). For diagnosing PV, the area under the curve (AUC) of NLR was better than that of EPO (0.921 vs. 0.827, P=0.003). Panmyelosis, which is essential for the diagnosis of PV, increases neutrophil count and decreases lymphocyte fraction. Consequently, patients with PV have a higher NLR than those with secondary polycythemia.
UNMET NEEDS IN CURRENT PV/ET TREATMENT
In addition to morbidity and mortality caused by thrombotic events and disease progression, individuals with ET and PV often have disease-related constitutional symptoms that may impact their quality of life. These symptoms include fatigue, pruritus, early satiety, abdominal discomfort, and weight loss. The symptomatologic etiologies of MPN also include direct treatment effects, cytopenia, splenomegaly, and disease biology. The MPN Landmark survey was conducted on 813 patients in the United States, wherein MF, PV, and ET were used to determine the effects of symptom burden on an individual’s quality of life [22]. While there was a significant correlation between symptom ratings and risk stratification in patients with MF, there was no such correlation in patients with ET and PV. Sixty percent of the patients with low-risk ET and PV had adverse effects impacting their quality of life, with fatigue being the most prevalent; 25–33% of low-risk patients reported missing at least one day of work in the preceding 30 days owing to symptoms. Similarly, an investigation of symptom clusters among patients with MPN revealed that a significant symptom load was also apparent in the low-risk disease groups [23]. To capture these symptoms directly from patients with MPNs, the Myelofibrosis Symptom Assessment Form was developed, which was subsequently modified to include ET and PV to form the Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF) [24, 25]. Numerous studies have shown that ruxolitinib and interferon (IFN) are effective in the symptomatic management of patients with MPNs [26, 27]. Complete responses were obtained in patients with PV and ET who were treated with either hydroxyurea (HU) or pegylated interferon alfa-2a (peg IFNα-2a); however, only 19–32% of the patients reported clinically significant improvement in symptom reduction [28]. Thus, the absence of a correlation between hematologic and symptomatic improvement shows that symptomatology should receive more attention.
Patients with PV and ET have lower overall survival (OS) than the general population [29]. In a cohort of 826 patients with MPNs at the Mayo Clinic, the respective median survival rates for ET, PV, and PMF were approximately 20, 14, and 6 yr, whereas the comparable values for patients <60 yr were 33, 24, and 15 yr, respectively [30]. It was also observed that survival rates declined with age in patients with MPNs, as determined by classifying the patients as follows: (1) under 40 yr of age; (2) 41–60 yr of age; and (3) above 60 yr of age [31]. Based on these investigations, young patients are often classified as low-risk and thus do not need to undergo active therapy. However, a recent study showed a considerable increase in excess mortality among younger patients with MPNs [32]. In this study, excess mortality was defined as the ratio of the observed mortality in patients with MPNs to the mortality predicted for patients of the same age. Particularly, excess all-cause mortality was greater among patients <60 as compared to those ≥60 in the ET (relative risk, RR 2.75 vs. 1.82; P<0.001) and PV (RR 3.16 vs. 1.92; P<0.001) groups. Furthermore, death rate also increases with age; however, the fact that the relative risk is higher in younger patients than in the general population indicates that they should receive more attention.
Patients with ET and PV are generally treated for a long period of time. HU is the standard treatment for high-risk patients with PV and ET. It also has the advantages of being effective, simple to administer, and affordable. It has been observed that long-term therapy with HU in patients with MPN is related to an increased incidence of secondary malignancies, thus raising concerns about its possible mutagenic impact [33]. Additionally, no therapy has shown a disease-modifying effect in randomized clinical trials. However, recent studies have demonstrated the disease-modifying potential of IFNα [34]. A retrospective single-center analysis of 470 patients with PV compared myelofbrosis-free survival (MFS) and OS in patients treated with recombinant IFNα (rIFNα) with HU or phlebotomy-only (PHL-O). Patients were categorized based on their first cytoreductive therapy received for at least 1 yr. In low-risk patients with PV (262 patients), the 20-yr MFS for rIFNα, HU, and PHL-O was 84%, 65%, and 55%, respectively (P<0.001); however, the OS results were not significant. In 208 high-risk patients with PV (208 patients), the 20-yr OS for rIFNα, HU, and PHL-O was 66%, 40%, and 14%, respectively (P=0.016); however, the MFS results were not significant. In the multivariable analysis, a longer time on rIFNα was associated with a lower risk for myelofibrosis (HR: 0.91, P<0.001) and lower mortality (HR: 0.94, P=0.012). Although this was a retrospective study and did not reflect treatment changes after the first year, it is significant because it was followed for a median of <10 yr.
For these reasons, there is growing interest in the active treatment of low-risk patients with PV and ET [35, 36]. The European Leukemia Net (ELN) 2021 recommendations suggest that low-risk patients with PV should begin cytoreductive drug therapy if at least one of the following criteria are fulfilled: (1) strictly defined intolerance to phlebotomy, (2) symptomatic progressive splenomegaly, (3) persistent leukocytosis, (4) progressive leukocytosis, (5) extreme thrombocytosis, (6) inadequate hematocrit control requiring phlebotomy, (7) persistently high cardiovascular risk, and (8) persistently high symptom burden. In these cases, rIFNα, either in the form of ropeginterferon alfa-2b (ropeg IFNα-2b) or peg IFNα-2a, is the recommended cytoreductive treatment for low-risk patients with PV [37].
Patients with PV and ET survive for a long time; the incidence of thrombosis, MF/leukemic transformation as well as the mortality remain low. A lengthy observation time is necessary to perform prospective research, thus rendering it difficult to accomplish. Therefore, it is necessary to actively develop new endpoints such as symptom improvement.
NOVEL AGENTS FOR PV/ET TREATMENT
Currently, INF and ruxolitinib are administered in addition to HU. Here, we present the most recent studies on INF, ruxolitinib, and new emerging agents.
Interferon
IFNα was the first immunotherapeutic drug approved for clinical use in cancer by the Food and Drug Administration (FDA) in 1986 [38]. It is a cytokine and is among the molecules utilized for cell-to-cell communication to activate the immune system. Initial research identified IFNα as an effective therapy for controlling thrombocytosis in MPNs [39, 40]. Since then, several studies have confirmed that IFNα can also inhibit myeloproliferation in MPNs, reduce the need for phlebotomies in PV, relieve pruritus, normalize elevated leukocyte and platelet counts, and reduce splenomegaly. Despite these benefits, IFNα is not widely used to treat MPNs because of its relatively high rate of discontinuation owing to numerous side effects. With the development of peg IFNα-2a and ropeg IFNα-2b, side effects have been reduced and administration intervals have been extended. Therefore, IFN is a promising treatment of choice for disease modification, especially given its impact on mutation burden [27].
Masarova et al. [41] described the findings of a single-center, prospective, phase II study of peg IFNα-2a in patients with ET (N=40) and PV (N=43), with a median follow-up of 183 months. At the time of the final follow-up, 41% of the patients (N=27) had a hematologic response, while 42% had a molecular response. The 15-yr follow-up of peg IFNα-2a in patients with ET and PV supports the durability of responses and disease control in patients who can tolerate long-term treatment with an acceptable level of safety.
The Myeloproliferative Disorders Research Consortium 112 was an investigator-initiated phase 3 trial comparing HU to peg IFNα-2a in treatment-naïve high-risk patients with ET (N=81) and PV (N=87) [37]. At 12 months, the complete response (CR) for HU was 37%, while that for peg IFNα-2a was 35% (P=0.80). At 24 to 36 months, the CR for HU was 17–20%, while that for peg IFNα-2a was 29–33%. Furthermore, grades 3-4 treatment-emergent adverse events were more frequent with peg IFNα-2a. Both agents effectively prevented thrombotic events and disease progression. Thus, IFN can also be considered as a first-line option for patients with ET/PV who require cytoreductive therapy.
Ropeg IFNα-2b is a mono pegylation IFNα with a prolonged half-life, thereby permitting dosing every other week. The non-inferiority phase III PROUD-PV/CONTINUATION-PV trial randomized 257 patients with PV who were either HU-naïve or HU-pretreated for <3 yr to ropeg IFNα-2b or HU [42]. The complete hematological responses in the ropeg IFNα-2b group versus the standard therapy group were 53 (43%) of 123 patients versus 57 (46%) of 125 patients (P=0.63) at 12 months (PROUD-PV), and 67 (71%) of 95 patients versus 38 (51%) of 74 patients (P=0.012) at 36 months (CONTINUATION-PV). Ropeg IFNα-2b showed a greater decrease in JAK2V617F allele burden over time as compared to that by HU. Recently, the long-term effectiveness and safety of ropeg IFNα-2b were established in the PROUD-PV/ CONTINUATION-PV trial [43]. A reduction in the JAK2V617F allele burden was also observed in patients treated with ropeg IFNα-2b; JAK2V617F allele burden <1% at 6 yr was achieved in 19/92 (20.7%) patients in the ropeg IFNα-2b arm with baseline allele burden >10%. One patient in the control arm met this threshold (1/70, 1.4%; P=0.0001). Over 6 yr of therapy, event-free survival (risk events: disease progression, death, and thromboembolic events) was substantially greater among patients treated with ropeg IFNα-2b than in the control group (risk events reported in 5/95 vs. 12/74 patients, respectively; P=0.04). Therefore, this was the first prospective trial showing that ropeg IFN-2b therapy improves event-free survival and reduces the burden of the JAK2V617F allele.
CONTINUATION-PV was analyzed by dividing the subjects into low- and high-risk groups [44]. Ropeg IFNα-2b was effective in both groups; however, low-risk patients may have demonstrated a larger potential benefit i.e., higher hematologic and molecular response rates may be achieved properly; these patients are more likely to adhere to long-term therapy. These findings provide further support for early cytoreductive treatment initiation, as previously described in the revised ELN recommendations [37].
Ruxolitinib
Ruxolitinib, a JAK 1/2 tyrosine kinase inhibitor, was reported to decrease the production of pro-inflammatory cytokines in malignant MPN clones [45]. In the RESPONSE-1/-2 study, it showed greater efficacy in terms of hematologic response in patients with HU intolerance or resistance with or without splenomegaly, as compared with the best available therapy [26, 46]. In particular, ruxolitinib has shown potential benefits in terms of symptom alleviation in patients with PV/ET [26, 46, 47]. Thus, ruxolitinib may be considered for patients treated using HU, who require a change in medication.
Rusfertide (hepcidin mimetics)
Phlebotomy is a treatment option for patients with PV having a hematocrit level ≥45% [48]. However, aggressive phlebotomy may worsen iron deficiency. Iron deficiency symptoms e.g., fatigue, leg cramps, and general weakness, are often seen in patients with PV showing normal hemoglobin levels and who undergo therapeutic phlebotomy [49]. In low-risk PV patients, high phlebotomy needs are often an indication that cytoreductive therapy should be initiated.
Consequently, modulation of iron metabolism has become a promising therapeutic target. Hepcidin acts as a negative regulator of the iron pathway, thus resulting in the downregulation of ferroportin expression and a decrease in serum iron and transferrin saturation. Rusfertide is a hepcidin mimetic designed to bind and internalize ferroportin. It decreases iron release from macrophages and iron absorption from food. Rusfertide therapy in various pre-clinical studies have shown a decrease in the hematocrit in erythrocytosis mouse models [50]. Hoffman et al. [51] reported the results of two phase 2 trials investigating the activity of rusfertide in PV patients. The first trial (NCT04057040) was conducted in phlebotomy-dependent patients with PV (≥3 phlebotomies in the 6 mo with or without concurrent cytoreductive therapy) with a hematocrit (HCT) <45% at study entry. This study comprised of the following: (1) a 28-week open-label dose-finding phase; (2) a 12-week double-blinded randomized (1:1) withdrawal; and (3) a 3-yr open-label extension with all subjects receiving rusfertide. Rusfertide doses of 10–120 mg were self-administered subcutaneously every week in addition to prior standard therapy and adjusted monthly to maintain HCT <45%. In Study 1, 63 participants were included. The mean number of therapeutic phlebotomy (TP) within 28 weeks prior to enrollment was 4.63 and was 0.43 following therapy. On rusfertide, patients consistently maintained an HCT <45%, essentially eliminating TP, and had normalized serum ferritin, mean corpuscular volume, and iron. Rusfertide-treated patients also reported a statistically significant improvement in the symptom burden at week 28. The second study (NCT04767802) enrolled patients with poorly controlled PV with HCT >48% at study entry, despite TP with or without hydroxyurea. The rusfertide dose was started at 40 mg twice weekly and decreased once weekly when the HCT was 45%. In Study 2, 20 participants were included. The mean HCT was 50.7% before therapy, while the mean time to attain HCT=45% without TP was 4.79 weeks; subsequently, HCT was consistently well-controlled. Rusfertide was well-tolerated, with mostly grade 1-2 adverse events (AE). The most common AEs were injection site reactions. These were typically transient, manageable with topical therapies and did not lead to withdrawal from the study. In patients with PV with sub-optimally managed erythrocytosis, the addition of rusfertide to conventional treatment showed significant efficacy. A double-blinded phase 3 study (verify) to add rusfertide or placebo to the ongoing therapy is currently in progress.
Givinostat (histone deacetylase inhibitor)
Givinostat is an orally bioavailable, potent inhibitor of class I and II histone deacetylases (HDACs). It is being evaluated for safety and efficacy in the treatment of Duchenne and Becker muscular dystrophy and PV. It acts directly on JAK2V617F mutated cells by downregulating JAK2 protein synthesis and subsequently inhibiting its downstream signaling, thus reducing their proliferation. Moreover, givinostat favors the development of non-mutated over mutated colonies, thereby suggesting that it can restore normal hematopoiesis in PV patients [52, 53]. In phase I/II clinical studies of PV, givinostat was well-tolerated and provided encouraging clinico-hematological results [54, 55]. However, the persistent low-grade toxicity of this drug makes its long-term administration challenging.
Bomedemstat (LSD-1 inhibitor)
Lysine-specific demethylase-1 (LSD1) is an epigenetic enzyme that is essential for malignant cellular renewal and hematopoietic differentiation. Bomedemstat is an orally active LSD1 inhibitor that is effective in lowering peripheral blood counts, splenomegaly, and inflammation. CTP-201 is an ongoing phase 2 trial for evaluating the safety, effectiveness, and pharmacodynamics of bomedemstat in patients with ET and who have failed at least one standard treatment [56]. Overall, 73 patients were enrolled in the study. Median time on treatment was 23 weeks (0.1–84). In patients treated for ≥24 weeks, 94% (34/36) achieved a platelet count response ≤400×109/L without new thromboembolic events and within a median time of 8 weeks. At week 24, 79% (11/14) of the patients with MPN-SAF TSS >20 at baseline (28/73) showed improvement. At week 24, 67% (N=24) of the allele frequencies had decreased. In the safety population (N=73), the most frequent adverse events were dysgeusia (43%), constipation (27%), tiredness (23%), thrombocytopenia (23%), arthralgia (21%), contusion (16%), and diarrhea (15%). Serious adverse events were much less frequent, and only two drug-related events were documented. Phase 3 research of bomedemstat for the treatment of ET is currently being conducted.
CONCLUSION
Patients with ET/PV had a higher life expectancy and a lower risk for thrombosis and disease progression than patients with other hematologic malignancies. Young patients without a history of thrombosis have been categorized as low-risk and have received comparatively little attention. However, a recent report indicated that these patients have a higher relative risk than older patients when analyzing excess mortality in consideration of age. Thus, the development of medications with the potential for altering disease progression coincides with the growing interest for actively treating low-risk patients. Additionally, patients with ET/PV who have lived for a longer period of time should be monitored for symptom improvement. In addition to traditional HU, IFN and ruxolitinib may be used according to certain circumstances. Therefore, further research on new medications is required.
 XML Download
XML Download