

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Primary cutaneous lymphoma (PCL) refers to a group of non-Hodgkin lymphoma (NHL) subtypes with unique characteristics, which specifically involve the skin at diagnosis [1]. Primary cutaneous T-cell lymphoma (CTCL), the disease entity of T-cell derived PCLs, accounts for 70–85% of PCLs, whereas B-cells predominate in other NHLs [2, 3]. A consensus classification of cutaneous lymphomas was established by the European Organization for Research and Treatment of Cancer (EORTC) and the World Health Organization (WHO) in 2005 [4] and then updated in 2018 [5]. Recently, the classification was further refined by the WHO and the International Consensus Classification (ICC) [6, 7].
CTCL is a rare disease, making up 4% of all NHLs [8]. In the United States, the incidence is 6.4–7.7 per million persons, according to the Surveillance, Epidemiology and End Results (SEER) database [9, 10]. Mycosis fungoides (MF) and Sézary syndrome (SS) are the most common subtypes, together comprising two thirds of CTCLs [4]. Both MF and SS have a male predominance, with a 2:1 ratio versus females. The median age at diagnosis is 55 years, with the incidence increasing with age. However, MF/SS can occur in younger individuals and even in children [11]. While the prognostic impact of age is not clear, ethnic differences have been reported, with a higher incidence in Blacks and non-Hispanics (11.5), followed by Hispanics (7.9) and Asian/Pacific Islanders (7.1) [12, 13]. A recent study showed that MF in Black patients is associated with a female predominance, younger age, and a worse prognosis [12, 13]. The incidence of CTCL in Korea is low, with an age-adjusted rate of 1.3 per million persons in 2012, based on the Korean National Cancer Incidence Database (KNCIDB) [14]. The estimated 5-year survival rate in all registered patients, between 2008 and 2012 was 87.7%, with a trend of a worse outcome according to age.
MF first presents in the skin, but in its advanced stage it can also involve the lymph nodes, other organs, or blood. Typical skin lesions are patches and plaques with various morphologic and pathologic findings. SS is a distinctive disease category defined by erythroderma and neoplastic T-cells typically involving the blood [4, 5]. It is regarded as a leukemic form of MF, with refractory symptoms and a poor prognosis.
Although some studies have suggested an association of MF or SS with environmental exposure to radiation and chemicals and with infections, especially Human T-lymphotropic virus type I (HTLV-I) [15-17], the causes of these two diseases are unclear. MF and SS are sporadic, but a few cases of familial MF/SS have been described, thus implicating genetic alterations in the development of MF/SS [18-20]. Accumulated knowledge on the cell of origin, genetic landscape, and immunologic changes has led to a better understanding of the pathogenesis of MF/SS and provided new insights into potential therapeutic targets [21-24]. Here we review the clinical manifestations, diagnosis, risk stratification, and treatment strategy of MF and SS, focusing on the recent advances in the management of MF/SS.
CLINICAL MANIFESTATION
The majority of patients with MF suffer from persistent, slowly progressive skin lesions. The most common and exhausting symptom is pruritus, occurring in over two-thirds of MF patients and significantly affecting their quality of life [25, 26], due to its association with insomnia, anxiety, and depression. Accordingly, skin-directed treatment is main approach to the management of early-stage MF.
The skin lesions in MF are heterogenous, varying in size and shape. They initially present as localized or generalized patches, plaques, and/or papules, with possible progression to tumor formation and generalized erythroderma (Fig. 1A–C, G). Patches/plaques/papules extending over a body surface area of <10% (T1) are seen in 30%, and more generalized manifestations (T2) in 35% of MF patients. A dome-shaped solid tumor ≥1 cm in diameter develops in 20% and erythroderma in 15% of patients, especially those with advanced disease [27]. Hypo/hyperpigmentation, alopecia, erosions, bullous lesions, and dry scaling are common accompanying findings. More than 30% of MF patients suffer from alopecia, which varies in its extent from patchy to generalized and in some patients is irreversible [28].
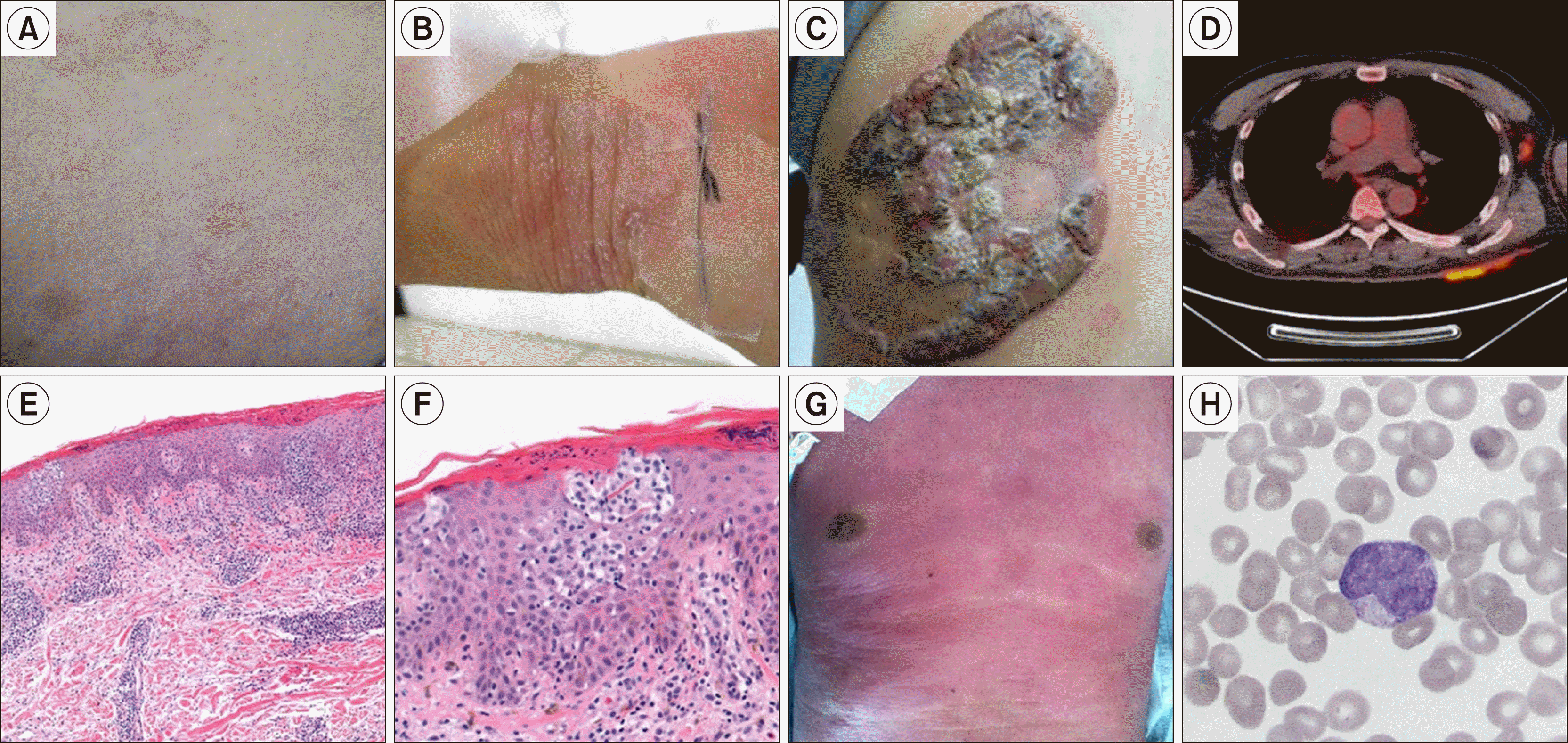
Fig. 1
Clinical manifestation and pathologic findings. Patchy (A), plaque (B), and tumor-type (C) skin lesions in a patient with advanced stage mycosis fungoides. PET scan showed FDG-uptake on large extent plaque and tumor stage lesions and axillary lymph nodes (D). Histology of plaque-type MF lesion with epidermotropic infiltration of lymphocytes, in which clonality of TCR gene rearrangement was confirmed (E). Higher resolution of E, representing grouped aggregation of large lymphocytes in the epidermis, forming early form of Pautrier’s micro-abscess (F). Generalized erythroderma in a patient with Sézary syndrome (G). Atypical lymphocyte with cerebriform nuclei, so called ‘Sézary cell’, was observed on peripheral blood smear (H).
![]()
The term Sézary syndrome originated with the 1938 publication of a landmark case series that identified lymphocytosis with typical grooved, cerebriform nuclei in patients with MF [29]. SS is a distinctive leukemic involvement of CTCL, characterized by generalized erythroderma but also sharing clinical and pathologic features with MF, as well as the same diagnostic approach and staging system [30]. However, SS is associated with more severe symptoms, a more unfavorable response to treatment, and worse survival than is MF. The skin lesions in SS are generally diffuse, rather than the progression from patches to plaques to tumors that occurs in MF. Lymphadenopathy and pruritus are frequent features of the disease.
The lymph nodes are the most common extracutaneous site and are involved in 30% of MF/SS patients [31]. Bone marrow involvement is rare, but peripheral blood involvement exclusively correlates with the extent of the skin lesions. Other extracutaneous lesions are rare but may develop in the lungs, spleen, liver, gut, and central nervous system, especially in patients with generalized erythroderma including those with SS [1, 32, 33].
DIAGNOSTIC APPROACH
The initial diagnostic work-up for MF should consist of a comprehensive physical exam, laboratory tests, including a complete blood cell count, routine chemistry including lactic dehydrogenase (LDH), and histologic confirmation. During the physical exam, the size, location, and degree of involved skin surface area should be recorded. To detect changes in skin lesions, standardized photographs are encouraged at baseline and at each subsequent assessment. Obtaining a representative skin biopsy is the most important step in the diagnostic work-up. Imaging studies such as computed tomography (CT) and fluorodeoxyglucose-positron emission tomography (FDG-PET) scans are required to evaluate extracutaneous disease (Fig. 1D). A bone marrow examination is not mandatory but is highly recommended.
The diagnosis of early-stage MF is challenging because the skin manifestations and biopsy findings may be misinterpreted as nonspecific and reactive changes. Many patients will have had other dermatologic presentations, such as nonspecific dermatitis, poikiloderma, or erythroderma and symptoms for years to decades before receiving a definitive diagnosis of MF [11, 27, 37]. For those patients, the clinical course should raise suspicion of MF even if the skin biopsy is non-diagnostic. Multiple skin biopsies, each consisting of at least 4-mm punch biopsy over the most indurated area are recommended. Topical treatment should be stopped for >2 weeks before the skin biopsy. The ISCL suggested diagnostic algorithm for early MF as shown in Table 1 [38].
Table 1
Diagnostic algorithm for early Mycosis Fungoides [38].
![]()
The characteristic histopathologic finding in a patch/plaque MF is an epidermotropic infiltration of neoplastic T cells (Fig. 1E, F). In early-stage, small cerebriform lymphocytes are observed in epidermis along the basal layer. When the disease progresses, denser band-like infiltration occurs, sometimes forming Pautrier’s micro-abscess, which are aggregates of tumor cells in the epidermis. It is not common, but they are specific for MF/SS. In tumor-stage, dense monomorphic infiltration of neoplastic lymphocytes involves the full thickness of dermis in nodular or diffuse pattern. In advanced stage MF and SS with blood involvement, lymphocytosis with typical grooved, cerebriform nuclei, so called Sézary cells, can be observed on peripheral blood smear (Fig. 1H).
There are some clinicopathologic variants of MF. Folliculotropic MF has a tropism for the epithelium of hair follicles, which often involves face and neck. Its manifestation includes a grouped papule, cysts, comedones, localized alopecia, and pseudo-tumors. Superficial form of this variant shows favorable survival similar to classic MF, however, a deep variant is associated with aggressive disease course and poor prognosis [39, 40]. Pagetoid reticulosis is a localized form with strong epidermotropism, generally involving distal areas of extremities [5]. It is usually indolent and can be treated with local radiation. Granulomatous slack skin is another rare variant of MF, which develops redundant skin slowly but progressively, typically in axillary and inguinal area. Histopathology of this variant shows dense infiltration of lymphocytes with granulomatous features and infiltration of atypical lymphocytes into the superficial layers with a variable distribution into the epidermis [5].
In MF/SS, malignant T-cells are predominantly CD4+, with a high CD4/CD8 ratio, and the aberrant loss of other pan-T-cell antigens, including CD2, CD3, CD5, and CD7. Immunohistochemical staining of biopsy specimens and flow cytometry of the peripheral blood can be facilitate the detection of clonal T-cells allowing a presumptive diagnosis of MF/SS. For SS, in addition to the morphological evaluation, multicolor flow cytometry is useful to detect Sézary cells, which are generally CD3+, CD4+ and CD8- [41]. The aberrant loss of other T-cell antigens, including CD2, CD3, CD4, CD5, CD7 and CD26, is a common finding, similar to the neoplastic cells found in MF [42-44]. The loss of CD7 or CD26 is sensitive and highly specific for SS [45].
Clonal gene rearrangement of the T-cell receptor (TCR) can be detected by PCR-based methods in formalin-fixed, paraffin-embedded tissues [46]. Its sensitivity is high, but it should be cautiously interpreted because it may not be specific for malignancy, as it is also detected in older adults and in patients with benign diseases. The presence of identical T-cell clones in multiple sites is considered as specific for MF/SS [47], but cases of MF without a detectable T-cell clone have also been reported [48]. Next-generation sequencing (NGS) is a highly sensitive and specific approach to assessing the clonality of T cells in MF/SS [49, 50]. It will be helpful to make a diagnosis of MF, however, NGS also cannot discriminate T-cell clonality in early-stage MF from reactive lymphocytes completely [51]. The clonality of neoplastic T-cells can also be confirmed by flow cytometric analysis for TCR-β chain variable region family members (TCR-Vβ), in addition to PCR-based analysis. An altered ratio of two segments (C1 and C3) in TCR-β chain constant region is a useful biomarker of α/β T-cell clonality [52, 53].
In endemic areas, assays aimed at the detection of HTLV-I virus should be performed to aid in the differential diagnosis. In patients with extracutaneous lesions, such as in the lymph nodes, tissue should be obtained from those sites as well. Lymph node involvement is assessed according to the Lugano classification [54].
STAGING AND RISK ASSESSMENT
The revised tumor, node, metastasis, and blood (TNMB) staging system was established for the staging of MF/SS and includes several revisions to the original TNM staging system [30, 54, 55]. It is based on findings in the skin (T1–T4), lymph nodes (N0–N3), viscera (M0–M1), and blood (B0–B2) as shown in Table 2. A recent update by the International Society for Cutaneous Lymphomas (ISCL), the United States Cutaneous Lymphoma Consortium (USCLC), and the EORTC includes the role of clonality testing in the determination of blood involvement and staging. Bone marrow involvement was also defined and added to the visceral staging. For clonality assessment, PCR-based detection of a TCR-γ/β gene rearrangement and NGS are recommended.
Table 2
Mycosis Fungoides Cooperative Group TNMB classification of cutaneous T cell lymphoma [55].
![]()
As discussed above, SS is a unique form of advanced MF, defined by the presence of generalized erythroderma (T4) and of >1,000/μL Sézary cells in the peripheral blood (B2). Staging is based on the findings in the lymph nodes and/or visceral involvement and ranges from stage IVA1 to stage IVB. MF is diagnosed in patients with lymph node and/or visceral lesion but no B2 blood involvement.
Survival outcomes according to the staging system are shown in Table 3 [56]. Stage I–IIA is considered representative of early- or limited-stage disease. In these patients, overall survival (OS) is prolonged, measured in decades. In fact, the survival of patients with stage IA disease is comparable to that of the age-matched healthy population [27, 37]. Late- or advanced-stage diseases, defined as stage IIB or higher, are associated with a worse outcome; the median survival is <5 years. In patients with stage IVB disease, the median OS is 1.4 years, and 5-year survival is only 18% [56]. A recent meta-analysis reported a similar OS [57], as improvements resulting from new treatment approaches have yet to be included in survival analyses.
Table 3
Clinical staging and prognosis of mycosis fungoides and Sézary syndrome [56].
![]()
For a specific variant, considering its unique clinical course with relatively better prognosis, an alternative staging system for folliculotropic MF has been proposed [39, 58].
Clinical prognostic indices for early- and advanced-stage diseases have been investigated. For early-stage disease, the cutaneous lymphoma international prognostic index (CLIPi) has been widely used. Adverse prognostic factors include age (>60), sex (male), type of skin lesion (plaques or folliculotropic), and nodal stage (N1/Nx) [59]. The 10-year OS is 90.3% for low-risk disease (0–1 factors) and 48.9% for high-risk disease (3–5 factors).
For advanced-stage disease, the Cutaneous Lymphoma International Consortium (CLIC) suggested prognostic index, which included age >60 years, stage IV, elevated LDH level, and large cell transformation (LCT) as independent prognostic factors associated with worse survival outcomes [60]. The presence of large cells, which is frequently CD30+, may be seen in patient with MF/SS. LCT, increased large cell count >25% of the infiltrated lymphocytes, may occur in advanced-stage MF/SS and is associated with an adverse prognosis [61-64]. The 5-year OS is 68% in low-risk patients (0–1 factors) and 28% in high-risk patients (3–4 factors).
In addition, molecular biomarkers have been widely studied for the heterogenous MF/SS. Gene expression analysis showed 17 genes including IL2RA, CCR4, STAT5A, and TOX were associated with risk of disease progression in MF/SS and could distinguish MF from SS [65]. Later validation study showed that T-plastin and Twist are useful for the differential diagnosis of SS/MF from reactive changes and KIR3DL2 was associated with short response duration [66]. Several microRNAs, such as miR-223, miR-214, miR-486, and miR155, have been reported that their expression was prognostic, but it should be validated further [67-70]. Recent advances in technical analysis, high-throughput and more detailed analysis have been enabled; constitutive activation of oncogenic signaling pathway such as JAK-STAT signaling, altered cell cycle regulation, and epigenetic remodeling [71]. Mutations in genes involving T-cell activation, apoptosis, NF-kB signaling, and DNA repair, and frequent copy number variations in driver mutations have been demonstrated [22]. These findings enabled to understand the pathogenesis of MF/SS better and gave new insights into new therapeutic strategy as well as precise diagnosis and risk assessment. Detailed list of suggested biomarkers for MF/SS are shown in Supplementary Table 1.
TREATMENT OF MF/SS
Treatment of early-stage MF
Early-stage (IA-IIA) MF is an indolent disease that primarily involves the skin. Treatment is focused on skin-directed therapies rather than systemic chemotherapy or immunotherapy, given the excellent survival outcomes of these patients. The main therapeutic goal for early-stage MF is the relief of symptoms (pruritus, pain, abnormal sensation, and cosmetic restoration). Less toxic but nonetheless effective treatments should be chosen to reduce the risk of disease progression. Thus, treatment should be determined according to disease extent, age, comorbidity, treatment availability, and safety profiles.
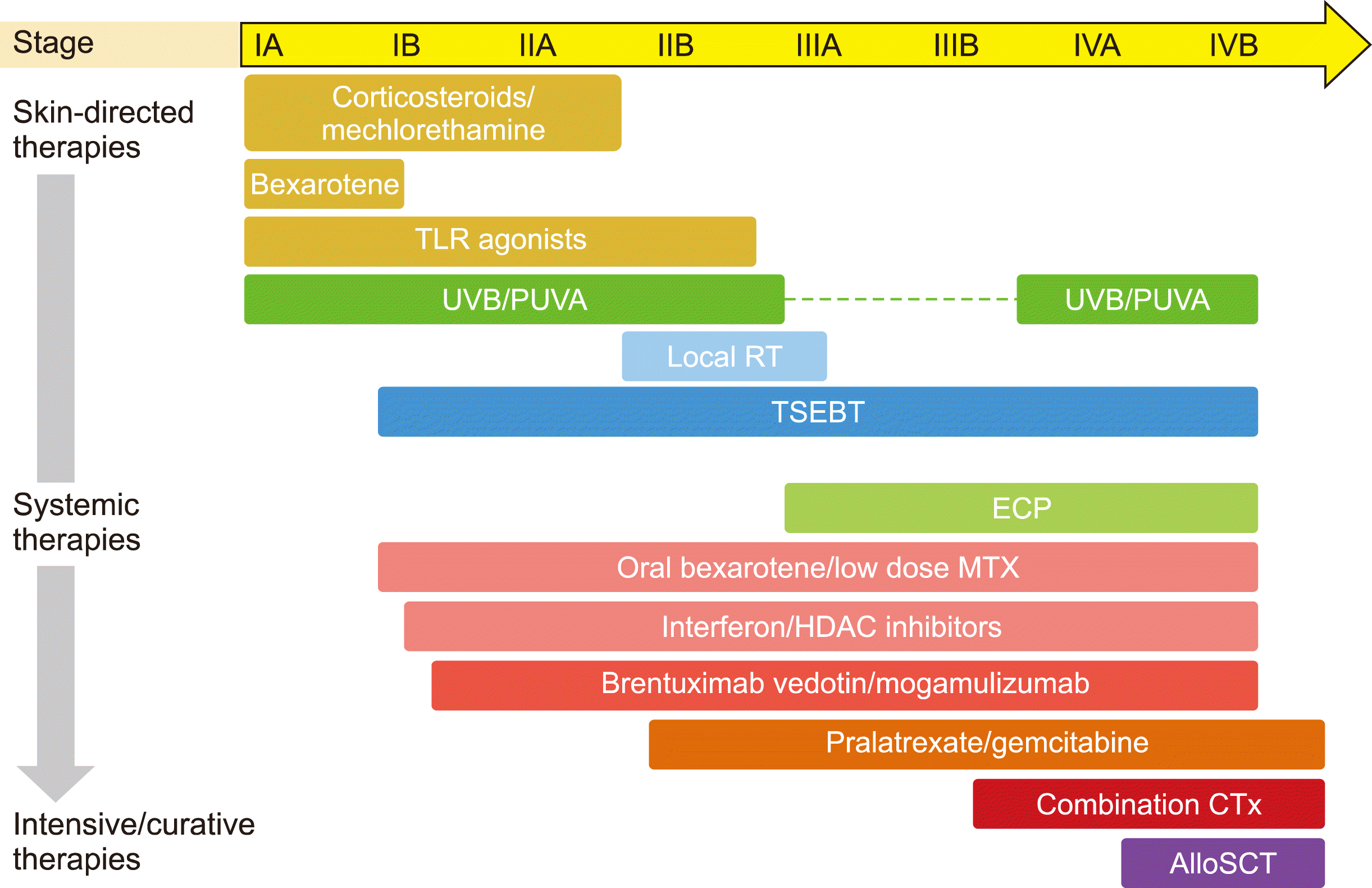
Skin-directed therapies include topical corticosteroids, retinoids, mechlorethamine, and imiquimod, localized radiation, total skin electron beam therapy (TSEBT), and phototherapy with the latter consisting of narrow band ultraviolet B (NBUVB) and psoralen with ultraviolet A (PUVA) (Fig. 2). Systemic therapies including oral bexarotene, interferon, immunomodulating agents, monoclonal antibodies, and targeted agents, can be used in patients with refractory disease and extensive symptoms. In this setting, drugs with a better safety profile are generally preferred, such as oral bexarotene and low-dose methotrexate.
The most common first-line therapy is topical corticosteroids, such as clobetasol propionate. Its efficacy was established based on retrospective reports of an overall response rate (ORR) of 95% for stage IA/IB disease and minimal toxicity [72, 73]. Topical mechlorethamine (0.02% gel) was evaluated in a phase II trial for stage IA-IIA MF, in which an ORR of 58.5% and a complete response rate (CRR) of 13.8% with some contact dermatitis and irritation were reported [74].
For refractory or persistent skin lesions, bexarotene (retinoic acid, 1% gel) can be used; its ORR in prospective trials ranged from 44 to 63% [75]. Topical bexarotene and other retinoids may cause excessive skin irritation when applied over large areas and are thus appropriate only for stage IA disease.
Topical toll-like receptor (TLR) agonists induce the production of cytokines, such as interferons, leading to anti-tumor immunity [76]. Studies of imiquimod, a TLR7 agonist, reported an ORR of 80% and a CRR of 45% [76, 77]. Fatigue and flu-like symptoms were reported as side effects but were rare. Clinical improvement and a reduction of neoplastic cells were also demonstrated for the TLR7/8 agonist resiquimod [78].
Generally, a topical corticosteroid, mechlorethamine, or phototherapy is recommended as the initial treatment. However, for patients with very severe symptoms and generalized, thick plaques, TSEBT will likely be more effective. In patients with disease relapse or failure to respond to one skin-directed therapy, another topical therapy or a combined strategy should be considered before systemic therapy is provided.
Phototherapy (UVB or PUVA), alone or in combination with other agents, is another effective skin-directed therapy. Either UVB or PUVA is indicated in patients with stage IA-IIA MF [79], but thick plaques or folliculotropic MF may be more responsive to PUVA because of its superior skin penetration. PUVA is also the first choice in patients of color, including Asians. A consensus guideline for phototherapy was published by the USCLC [80].
For localized MF refractory to topical agents, local radiation may be considered, especially in patients with a single skin lesion. In patients with widely distributed lesions, TSEBT using low-dose radiation may be effective [81].
Treatment of advanced-stage disease
The goal of therapy in patients with advanced-stage disease is not only symptom relief and long-term disease control but also the prolongation of survival. The treatment modality should be chosen based on the goals of therapy at different time points during the disease course (Fig. 2). For long-term disease control, agents without cumulative toxicities or immunosuppression are a good choice whereas the treatment of life-threatening disease requires curative but more toxic therapies.
For patients with localized skin involvement, skin-directed therapies can be applied with or without systemic therapy. In those with extensive skin involvement or visceral involvement, systemic therapy is required. TSEBT in combination with systemic therapy is preferred for generalized diseases, however, its use in patients with erythroderma may be contraindicated because of the risk of severe desquamation [81].
Systemic therapy ranges from immunomodulatory drugs (bexarotene, interferon, and low-dose methotrexate) to targeted agents, such as histone deacetylase inhibitors (vorinostat and romidepsin), monoclonal antibodies (alemtuzumab), antibody-drug conjugates (brentuximab vedotin and mogamulizumab), immune checkpoint inhibitors (pembrolizumab), and other investigational drugs. Data on commonly used systemic therapies are provided in Table 4. Most traditional agents have been used based on the results of phase II or retrospective studies; consequently, their true efficacies are often unclear. Novel agents targeting the unique biology of MF/SS have been recently introduced. Their superior efficacy over historical systemic therapies has been demonstrated in randomized phase III trials. With increasing therapeutic options, the decision regarding the optical treatment approach has become more complicated.
Table 4
Systemic therapies for refractory or advanced-stage mycosis fungoides and Sézary syndrome.
| Reference | Agents | Phase | Prior Txa) | N | Outcomes | Comments |
|---|---|---|---|---|---|---|
| Duvic et al. 2001 [82] | Bexarotene | II/III | 2 | 56 | ORR 45% (CRR 2%) | Pancreatitis, hypertriglyceridemia, thyroid dysfunction |
| Zinzani et al. 2000 [103] | Gemcitabine | II | 3 | 44 | ORR 70.5% (CRR 11.5%) | Myelosuppression, elevated hepatic enzyme |
| Dummer et al. 2012 [102] | Peg-L-doxorubicin | II | 2 | 49 | ORR 40.8% (CR 6.1%) | Myelosuppression, gastrointestinal toxicity |
| Duvic et al. 2007 [93] | Vorinostat | II | 5 | 33 | ORR 24.2% (CRR 0%) | Fatigue, diarrhea, nausea, thrombocytopenia, abnormal echocardiogram |
| Whittaker et al. 2010 [96] | Romidepsin | II | 4 | 96 | ORR 38% (CR 5%) | IB-IVA |
| GI disturbances, asthenic conditions, ECG changes | ||||||
| Prince et al. 2017 [99] (ALCANZA) | Brentuximab vedotin vs. MTX or bexarotene | III, RCT | 3 | 131 | ORR4 50% vs. 10% | CD30+ (≥10%) MF (SS excluded) |
| PFS 16.1 vs. 3.5 M | Peripheral neuropathy in 66% (9% Gr3), discontinuation in 14% | |||||
| Kim et al. 2018 [94] (MAVORIC) | Mogamulizumab vs. vorinostat | III, RCT | 186 | ORR 28% vs. 5% (68% in blood) | Stage IB-IVB MF/SS (LCT excluded) | |
| Infusion related reaction, thrombocytopenia, drug eruption, discontinuation in 7% | ||||||
| Khodadoust et al. 2020 [101] | Pembrolizumab | II | 4 | 24 | ORR 38% (CRR 8%) | IIB-IV |
| Cutaneous flare reaction (without discontinuation) |
![]()
For MF and SS, the response to treatment should be assessed based on overall skin, nodal, visceral, and blood responses. In early clinical trials and retrospective reports, response criteria and outcome endpoints were not uniformly defined. In 2011 and 2022, the ISCL/USCLC/EORTC published consensus recommendations for clinical end points for MF/SS (Supplementary Table 2) [54, 55].
The systemic use of bexarotene, an oral retinoid-X receptor-selective retinoic acid, was first approved by the FDA in 1999, prior to the development of the topical agent. In a multicenter phase II/III trial, patients who received 300 mg bexarotene/m2/day had an ORR 0f 45% [82]. Common toxicities included hypertriglyceridemia, pancreatitis, and hypothyroidism and were dose dependent. Treatment for >6 months without adjuvant therapy, until disease progression if the drug is well-tolerated, is recommended [83].
Interferon α-2b or γ-1b is another immunomodulatory agent used in the treatment of MF/SS. An ORR of 50–70% and a CRR of 20–30% have been reported [84, 85]. In patients with early-stage disease, interferon can be a second-line option, and in those with advanced-stage disease, including tumor-stage lesions, a first-line option. Interferon can also be successfully combined with other skin-directed therapies, such as PUVA, bexarotene, and extracorporeal photopheresis (ECP) [86], a type of phototherapy in which psoralen exposure precedes extracorporeal circulation. Psoralen binds to DNA after UVA radiation and leads to apoptosis of lymphocytes as well as an enhanced host immune response induced by monocyte activation and dendritic cell differentiation [87, 88]. Following a landmark report of a response in 73% of patients, ECP has become a first line treatment for advanced-stage MF and SS [89]. A favorable response around rate of 60% and a durable response of >8 years in patients with a complete response have been reported [90, 91]. The median time to response is 6 months, but the long-term use of ECP may be associated iron deficiency. ECP can also be combined with other therapies.
Histone deacetylase (HDAC) inhibitors have an established safety profile, and their potential efficacy has been demonstrated in phase I studies for T-cell lymphomas, including some CTCLs [92]. A phase II trial for vorinostat showed an acceptable and durable response (ORR of 30% for a median 185 days in patients with advanced-stage disease) [93]. However, in the MAVORIC trial, vorinostat was inferior to mogamulizumab [94]. Romidepsin, evaluated in phase II trials, resulted in an ORR of 38% that was durable for >1 year in advanced-stage disease [95, 96]. With maintenance at a lower dosing schedule, responders achieved a durable response of 7–34 (median 15) months [97]. For both HDAC inhibitors, QT prolongation and cardiac toxicity are potential complications and should be monitored regularly [98].
Based on the phase III ALCANZA and MAVORIC trials, both BV and mogamulizumab were approved by the FDA, for patients with relapsed or refractory disease [94, 99]. Brentuximab vedotin (BV) and mogamulizumab are novel agents that have been evaluated in randomized phase III trials. In the ALCANZA trial, the efficacy and safety of BV compared with physician’s choice of therapy (methotrexate or bexarotene) were investigated [99]. The ORR4 (lasting at least 4 mo) and progression-free survival (PFS) were 50% and 16.1 months in the BV group vs. 10% and 3.5 months in the control group. After 1 year, 34.5% of patients in the BV group but 86.6% of those in the control group required next-line treatment. The efficacy of BV was observed at any level of CD30 expression and was independent of the presence of LCT. Mogamulizumab is a humanized monoclonal antibody for the chemokine receptor 4 (CCR4) that enhances antibody-dependent cell-mediated cytotoxicity and inhibits Treg-mediated immune suppression, leading to anti-tumor activity against MF/SS. In the above-mentioned MAVORIC trial, a randomized phase III study, mogamulizumab was tested for its efficacy in patients with relapsed/refractory CTCL and resulted in a better ORR and PFS than achieved in patients treated with vorinostat (28% vs. 5% and 7.7 vs. 3.1 mo) [94]. A favorable response in the blood compartment, frequently involved in SS, was evidenced by an ORR of 68%. The side effects of mogamulizumab included drug eruptions, which were mostly tolerable and not a cause of drug discontinuation.
Clinical features may guide the choice of a systemic therapy in patients with specific disease phenotypes. Romidepsin or mogamulizumab may be beneficial for patients with a high blood burden. The reported response rate in the blood compartment was 54% and 68%, respectively [94-96]. The median response duration in the subgroup of mogamulizumab treated patients with a blood response was 26 months. There is as yet no evidence of the efficacy of BV in the blood compartment, such that SS patients were excluded from the ALCANZA trial. Because patients with generalized erythroderma frequently have blood involvement, a similar strategy may be followed. For lymph node involvement, romidepsin, pralatrexate and BV are effective, as shown in clinical trials that included patients with CTCL and peripheral T-cell lymphoma. In the ALCANZA study, the extracutaneous response rate at 4 months was 46% in the BV-treated group vs. 9% in the control group [99]. However, in the MAVORIC trial, the nodal response rate to mogamulizumab was 17% [94]. LCT is a significant adverse factor in MF/SS patients and should be considered in the selection of systemic therapy. Although CCR4 is commonly expressed in transformed cells, mogamulizumab failed to show activity in patients with LCT in early-phase trials [100]. Thus, in the phase III MAVORIC trial, patients with LCT were excluded [94].
The feasible activity of immunomodulatory agents against MF/SS and the observation of high PD-L1 expression in some CTCLs have raised interest in immune checkpoint inhibitors. In a phase II trial of pembrolizumab [101], the ORR was 38%. However, the role of checkpoint inhibitors in the management of MF/SS has yet to be fully investigated.
For refractory and aggressive disease, cytotoxic chemotherapy, such as gemcitabine and liposomal doxorubicin, can be considered for disease control [102, 103]. Patients with very aggressive disease and thus with a reduced survival, such as those with LCT or B2 disease, will require curative agents that provide a rapid and strong response. The treatment strategy developed for aggressive T-cell lymphomas may be effective.
Despite the introduction of novel agents, allogeneic stem cell transplantation (SCT) remains the only curative modality in patients with advanced-stage MF/SS, although the data are thus far limited. Based on registry data, the 5-year PFS and OS were 17% and 32% with a 1-year non-relapse mortality (NRM) of 19% [104]. Similar results were reported in a systematic review and meta-analysis, in which the relapse rate was 47% and the NRM 19% [105]. Earlier transplantation in patients with CR1/CR2 or in those with relapse after a maximum of three systemic therapies were associated with a better outcome [106]; however, the optimal timing of allogeneic SCT is unclear yet. High-risk patients with an expected survival of <5 years may be a candidate for allogeneic SCT. As a bridging therapy, BV can be used without increasing pre- and post-transplant toxicities [107]. However, mogamulizumab should be considered cautiously because of its association with severe acute graft-versus-host disease as well as a report of treatment-related/non-relapse mortality in a study of other types of lymphoma [108].
CONCLUSION
MF/SS is a rare but distinct disease entity that is highly heterogenous with respect to its clinical features and prognosis. Recent progress in understanding the pathogenesis of MF/SS has contributed to advances in the diagnosis, staging, and risk assessment of MF/SS as well as to the development of novel therapies. However, with accumulating data obtained from studies of novel drugs, decisions regarding the optimal form of therapy and its timing are becoming increasingly complex. The efficacy and safety of combination therapies should be investigated further. Well-designed clinical trials are warranted and the participation of patients in those trials is encouraged.
 XML Download
XML Download