

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The proportion of chronic lymphoproliferative disorders caused by large granular lymphocyte (LGL) leukemia varies between 2% and 5% in North America and Europe, and from 5% to 6% in Asia [1]. T-cell LGL (T-LGL) leukemia accounts for approximately 85% of cases, whereas chronic NK-cell lymphocytosis is estimated to affect 10% of patients. In T-LGL leukemia, cytotoxic (CD8+) T cells proliferate clonally and clinically, manifesting as neutropenia, anemia, and/or thrombocytopenia along with lymphocytosis [2], and the disease may occur in conjunction with various autoimmune conditions [3]. Although our understanding of the pathogenesis of the disease is poor, recent advances in molecular research have shed some light on the underlying mechanisms [4]. Current treatments involve the use of immunosuppressive medications; however, as these do not provide satisfactory long-term outcomes, targeted therapies are the next step toward a cure.
CLINICAL FEATURES
At the time of diagnosis, one-third of patients present with asymptomatic cytopenia or autoimmune disorders. Recurrent infection caused by chronic neutropenia dominates the initial presentation. B symptoms are rare in LGL leukemia as it is an indolent disease, although fatigue and B symptoms may occur in 20–30% of cases. Splenomegaly occurs in 25–50% of cases, whereas hepatomegaly and lymphadenopathy are very rare [5, 6]. According to the two largest series [7, 8], the average T-LGL count is 1.7×109/L. The incidence of transfusion-dependent anemia varies from 6% to 22%, whereas pure red cell aplasia (PRCA) occurs in 8% to 19% of patients. The incidence of thrombocytopenia is low and has been described in fewer than 20% of cases. It is estimated that 50% of patients have neutropenia and that approximately 20% have severe neutropenia. Recurrent oral aphthous ulcers are often observed in neutropenic patients; however, some patients remain asymptomatic. Approximately 15–39% of patients develop infections secondary to chronic neutropenia, and severe septic complications result in death in approximately 5–10% of patients. No correlation appears to exist between the levels of T-LGL infiltration and the severity of cytopenia or systemic symptoms, even though T-LGL infiltration has been detected in over 70% of cases.
A notable association between T-LGL leukemia and autoimmune diseases (15–40% according to the series) has been observed [9]. Rheumatoid arthritis (RA), which occurs in approximately 15% of patients, is usually diagnosed before T-LGL leukemia becomes apparent. A positive rheumatoid factor score (40–60%), antinuclear antibody score (40%), and antineutrophil antibody score (20–60%) reinforce the immune context of this lymphoproliferative disease [10]. Additionally, Sjögren’s syndrome, autoimmune thyroid disorders, coagulopathy, and inclusion body myositis have been reported in some cases. Patients with LGL leukemia have been reported to experience vasculitis with cryoglobulinemia and pulmonary artery hypertension.
DIAGNOSIS
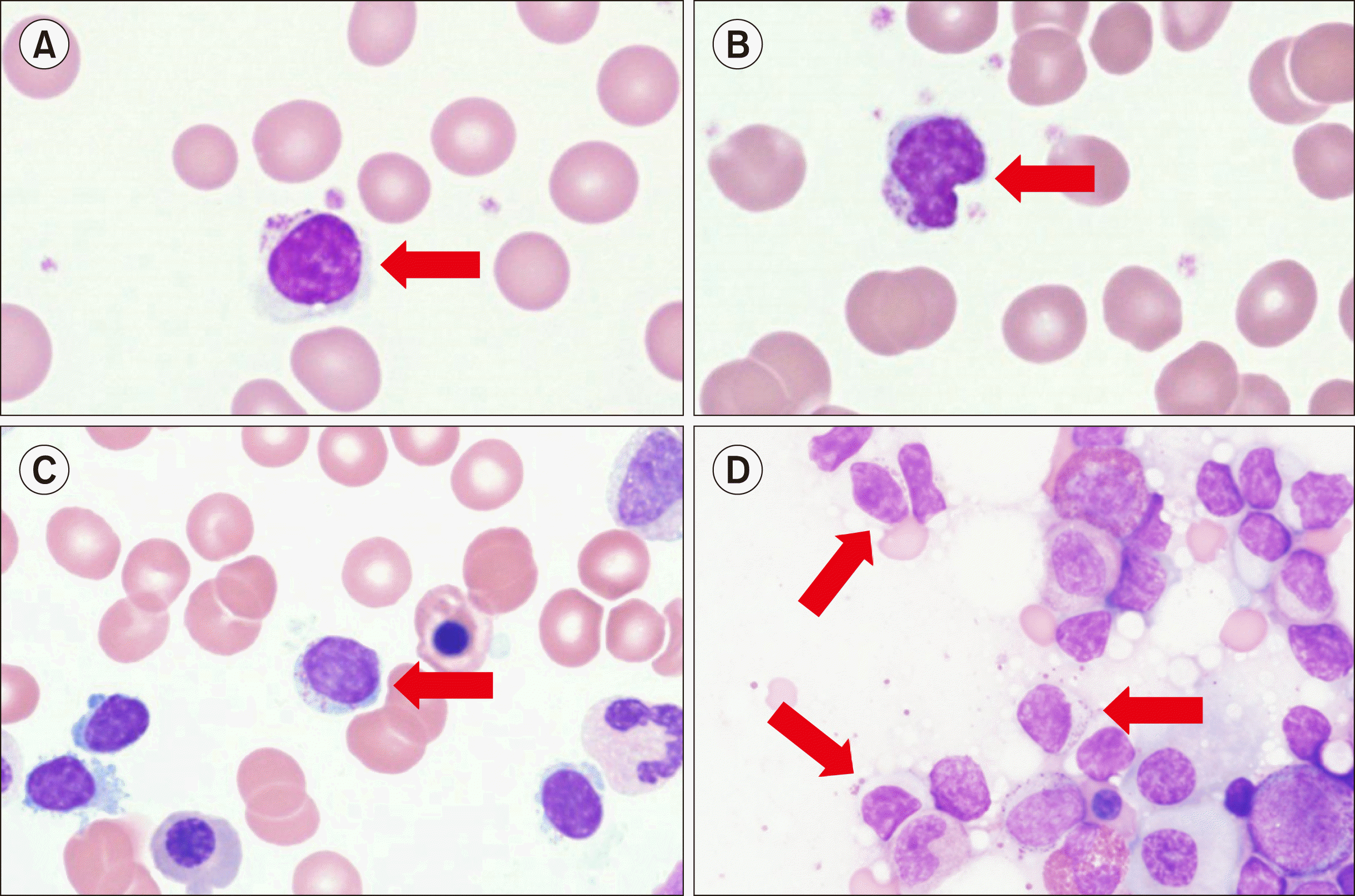
A diagnosis of T-LGL leukemia is suspected when the number of LGLs with moderate to abundant cytoplasm containing fine to coarse azurophilic granules in peripheral blood (PB) is more than 2×109/L. However, lymphocytes found in T-LGL leukemia are difficult to distinguish morphologically from activated cytotoxic lymphocytes observed in conditions such as infections and autoimmune diseases. Therefore, implementation of molecular methods to confirm the clonality of T-LGL cells is critical in the differential diagnosis of T-LGL leukemia from other benign conditions. Polymerase chain reaction using a probe for T cell receptor (TCR)γ is the preferred method for diagnosis of T-LGL leukemia, with flow cytometric analysis against Vβ TCR cell surface protein being a less sensitive method [11]. In a clinical setting, when T-LGL leukemia is suspected after morphological evaluation of PB, flow cytometric analysis is performed to confirm whether the immunophenotype of LGL cells found in PB is concordant with those found in typical T-LGL leukemia. T-LGL cells typically exhibit a mature post-thymic phenotype, and, in most cases, display CD3+, TCRαβ+, CD8+, CD16+, CD45RA+, and CD57+ and are CD4-, CD5dim, CD27-, CD28-, and CD45RO- [12]. Abnormal loss of expression of CD5 and/or CD7 is common in T-LGL leukemia, and expression of CD57 and CD16 occurs in over 80% of T-LGL leukemia cases [13]. Uncommon variants include CD4+/TCRαβ+ and TCRγδ+. Although bone marrow (BM) studies are not frequently performed in patients with T-LGL leukemia because identification of LGL cells in PB is not difficult, BM studies can be considered when the differential diagnosis is challenging. The typical BM infiltration pattern of T-LGL leukemia is hypercellular marrow with CD3+, CD8+, and CD57+ LGL cells in the BM, as observed using immunohistochemical stains [12]. The extent of BM involvement is variable, but usually constitutes less than 50% of cellular contents with interstitial/intrasinusoidal infiltration patterns. Because this pattern is difficult to identify through morphological evaluation, immunohistochemical staining against BM biopsy is useful for the sensitive detection of T-LGL cells with CD3+, CD8+, and CD57+ [13]. A case of T-LGL with PB/BM morphology is shown in Fig. 1.
PATHOGENESIS
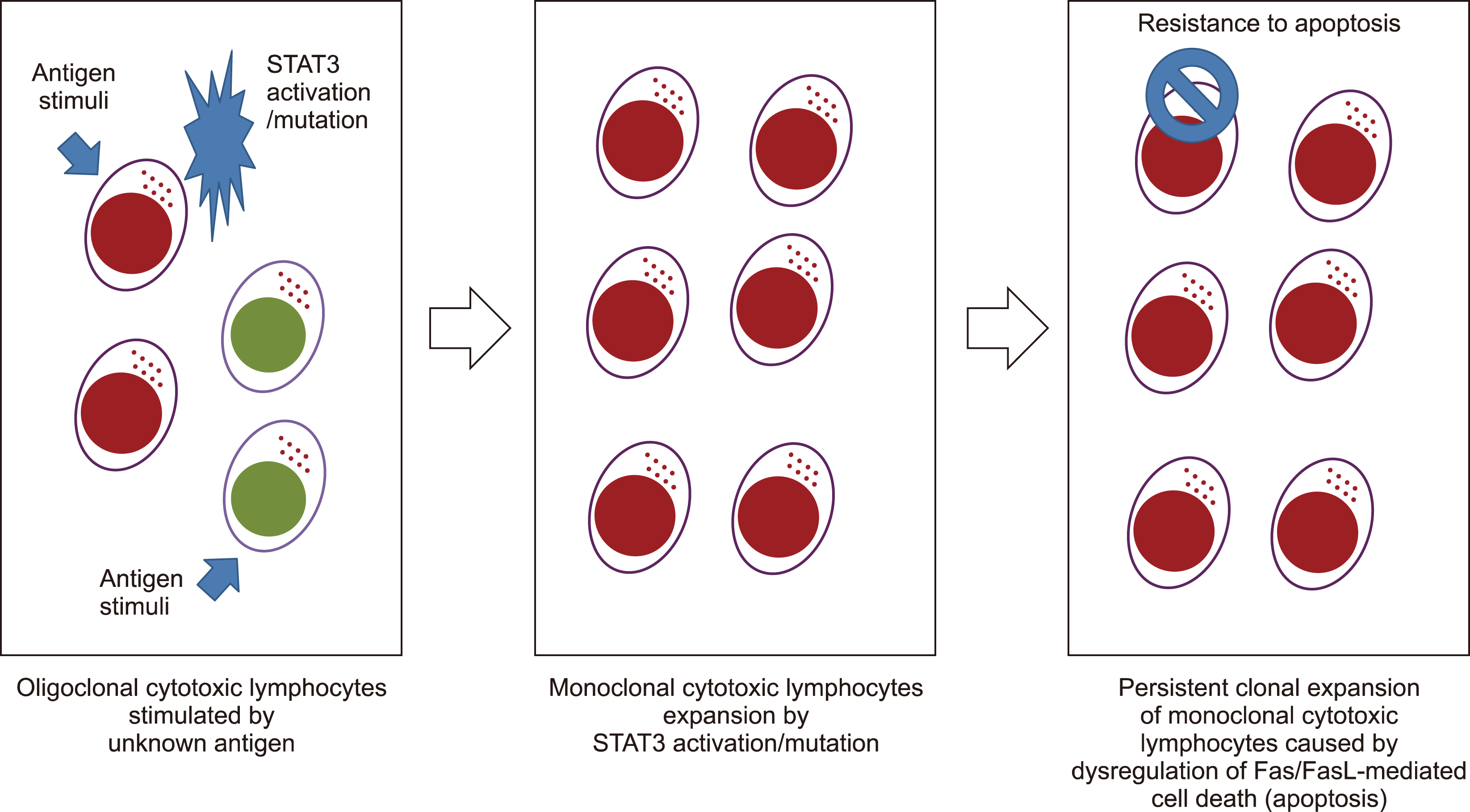
T-LGL leukemic cells exhibit a terminal effector memory phenotype, suggesting that they were first chronically stimulated by the immune system. In pathobiology, abnormal activation of the JAK/STAT pathway promotes survival and sustains the abnormal persistence of clones that lack effector functions [14]. Approximately 40% of patients with LGL have STAT3 sequence alterations [4]. As a result of somatic activation mutations, the leukemic cell population can expand and evolve more rapidly through enhanced activation from physiological stimulation [15]. STAT3 activating mutations modulate the transcription of anti-apoptotic Mcl-1, which in turn drives T-LGL expansion [14]. An increased level of IL-6 is observed in patients with T-LGL leukemia owing to the activation of the JAK-STAT3 pathway by the proinflammatory cytokine IL-6 [16]. Because SOCS3 transcription is induced by IL-6 via pStat3, it has been hypothesized that T-LGL leukemia has high levels of SOCS3 [16]. Moreover, the proinflammatory cytokine IL-15 and platelet-derived growth factor promote T-LGL survival and expansion, and IL-15 inhibits cell death by mediating pro-survival signaling [17]. In addition to altering Mcl-1 expression, IL-15 promotes the degradation of pro-apoptotic BID [18]. S1P, a pro-survival sphingolipid, is predominant in LGL leukemia, according to analyses of the molecular expression profile [19]. Owing to the increase in SphK1, which converts sphingosine into S1P, leukemic LGLs are more likely to undergo apoptosis when SphK1 is inhibited. LGL leukemia is associated with increased expression of S1P receptors, particularly S1PR5, which is activated by binding to S1P receptors through ERK1/2 signaling [20]. A diagram summarizing the pathogenesis of T-LGL is shown in Fig. 2.
TREATMENT
General concept of therapy
As the illness progresses, T-LGL leukemia is regarded as a condition that requires ultimate therapy. In fewer than 10% of patients with T-LGL, severe infection is the leading cause of disease-related mortality [7, 11, 21]. Ten-year survival is approximately 70%, and the majority of disease-related deaths result from severe infections. Patients should be frequently followed up for symptoms and disease development, regardless of treatment status, after the diagnosis has been confirmed. A “watchful waiting” approach and supportive treatment with antibiotics, erythropoietin, and/or granulocyte colony-stimulating factor (G-CSF) might be recommended. However, the use of G-CSF in patients with T-LGL may exacerbate splenomegaly or articular symptoms [22], while erythropoietin has been used to treat anemia caused by immunosuppressive medication [6]. An absolute neutrophil count of less than 500/µL, recurrent infections, transfusion-dependent anemia, or autoimmune diseases such as RA needing medication are indications for treatment [6, 12]. Because leukemic LGLs are constitutively activated cytotoxic cells, immunosuppressive medication is the cornerstone of T-LGL leukemia treatment. At least four months of treatment are necessary before evaluating the response.
FIRST-LINE THERAPY
Single oral immunosuppressive drugs, such as methotrexate (MTX), cyclophosphamide, or cyclosporine, are the traditional first-line treatment choices. MTX has been advocated as a first-line therapy, particularly for neutropenic patients [23, 24], based on prospective studies on its effectiveness. MTX is also the therapy of choice for primary RA; therefore, its usage is highly advantageous for patients with LGL having RA as an associated condition. Lamy and Loughran [6] reported a 55% overall response rate with a 21-month duration of response to MTX in a large group of 62 patients. A French series reported recurrence rates of 67%, indicating that the relapse rate may be significant [7]. Patients with STAT3 mutations and T-LGL leukemia are more likely to respond to MTX therapy and have a positive prognosis [24]. This study also showed that the total response rate to MTX was 38%, including both partial and full responders. Although MTX (10 mg/m2/wk) is typically well-tolerated, hepatic and pulmonary function of patients must be maintained as long as MTX treatment remains responsive.
Cyclophosphamide (50–100 mg/day orally) has been favored in the treatment of individuals with predominant anemia, especially PRCA or predominant anemia [25]. A retrospective analysis of 45 patients reported an overall response rate of 71% [26]. Because the response rate of cyclophosphamide is equivalent to that of MTX, it might continue to be used as a first-line treatment for T-LGL leukemia. Owing to the accompanying toxicity and increased risk of developing myelodysplasia or acute myeloid leukemia, cyclophosphamide should not be administered for more than 12 months [27, 28].
Cyclosporine (orally 3 mg/kg per day) is often reserved as an alternate first-line salvage therapy for patients in whom MTX and/or cyclophosphamide treatment fails. Response rates to cyclosporine vary with research; however, individuals with PRCA seem to have a favorable response [29]. However, if patients are responsive, it must be continued permanently, as discontinuation causes rapid recurrence of symptoms [6]. This is because clinical responses often occur without elimination of the leukemic LGL clone [30, 31]. Battiwalla et al. [32] showed that HLADR4 (present in 32% of LGL leukemia and 90% of RA cases) was a strong predictor of cyclosporine response.
SECOND-LINE THERAPY OR MORE
Owing to the rarity of the condition and the overall lack of prospective data, it is challenging to establish recommendations for patients resistant to first-line therapies. Purine analogs (fludarabine, cladribine, deoxycoformycin, and bendamustine) are recommended for resistant diseases, and were found to provide encouraging outcomes (79% overall response rate) in a small number of patients [33, 34]. To reduce toxicity, patients may be treated for a maximum of one to three cycles to elicit a rapid response. The combination of fludarabine and mitoxantrone has resulted in long-lasting full remission [33]. Young patients with refractory T-LGL leukemia may be candidates for hematopoietic stem cell transplantation. In a study of 15 patients with LGL leukemia who received autologous or allogeneic stem cell transplantation, six individuals remained disease-free after transplantation [35]. Patients with symptomatic splenomegaly may be candidates for splenectomies irrespective of the presence of cytopenia. A comprehensive literature analysis revealed an ORR of 56% without sustained responses [36].
POTENTIAL TARGET THERAPY
Considering the pathophysiology of LGL leukemia, specific inhibitors were tested in patients with the disease. Regarding IL-15 targeting, Waldmann et al. [37] demonstrated the effects of Hu-Mikb1, which inhibits IL-15 transpresentation to T cells that express IL-2/IL-15Rb (CD122), in a phase I trial. Hu-Mikb1 was found to be safe for patients, but no clinical reactions were detected. JAK/STAT3 inhibition might be a potential treatment strategy for patients with LGL leukemia. Weekly low-dose MTX has been shown to be a very effective inhibitor of the JAK/STAT pathway, and may explain its effectiveness in LGL leukemia [38]. However, STAT3-mutated subclones are still found in full responders, and the JAK3 pathway is now considered a target for immunosuppression [39]. Tofacitinib citrate (CP690550), a JAK3-specific inhibitor, has shown remarkable efficacy in the treatment of refractory RA [4, 40]. This particular inhibitor was evaluated in nine patients with refractory LGL leukemia and RA, six of whom showed a hematologic response, and neutropenia improved in five of the seven patients [40]. The novel multi-cytokine inhibitor BNZ-1 might be beneficial for patients with LGL leukemia. BNZ-1 inhibits the signaling of IL-2, IL-15, and, to a lesser extent, IL-9, but has no effect on IL-4, IL-7, or IL-21 [41]. In a phase I dose-escalation trial, BNZ-1 was demonstrated to be a highly active selective immunomodulator that decreases T regulatory cells, NK cells, and memory T cells while leaving the major leukocyte populations unaffected. In a phase I/II trial, it will be evaluated in patients with LGL leukemia (NCT03239392).
CONCLUSION
The complicated etiology of T-LGL leukemia, which is considered to include neoplastic, viral, and/or autoimmune processes, is characterized by a combination of molecular and systemic diseases. Although the role of Stat3 activation/mutation in the pathogenesis has led to significant advancements in our knowledge of LGL clonal proliferation, T-LGL leukemia is still an incurable condition and there is an unmet need to develop better therapeutic approaches.
 XML Download
XML Download