

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The BCR-ABL1-negative myeloproliferative neoplasms (MPNs) are a heterogeneous group of clonal hematopoietic neoplasms that include polycythemia vera (PV), essential thrombocythemia (ET), and overt primary or post-ET/post-PV myelofibrosis. Hyperactivation of signal transduction pathways, such as JAK/STAT, is a pathological hallmark of MPNs, resulting in increased numbers of myeloid lineage blood cells and systemic proinflammatory conditions. Myelocytosis and inflammation significantly increase the risk of vascular events of both arterial and venous origins. In addition, PV and ET can progress to secondary acute myeloid leukemia either directly or via transformation to myelofibrosis. In addition, some patients with overt primary myelofibrosis (PMF) eventually develop secondary acute myeloid leukemia (AML), which has a poor prognosis. MPNs are most common in patients in their 50s or above, although they can affect all age groups. PV and ET have indolent clinical courses, and the prevention of vascular events is a short-term treatment goal. In cases of overt myelofibrosis, patients experience systemic constitutional symptoms and splenomegaly as the disease progresses, and ruxolitinib and other JAK inhibitors are the current treatment standards. Some selected patients with a higher risk of myelofibrosis can be cured through allogeneic hematopoietic stem cell transplantation (allo-HSCT).
Similar to other areas of oncology, knowledge regarding the diagnosis and treatment of MPNs is rapidly evolving. In particular, by analyzing the genetic information of patients with MPN and associating this information with clinical variables, the survival and treatment outcomes of individual patients with MPN can be predicted much better than before. Here, we review several currently established and suggested prognostic systems and recommendations for patients with MPN, focusing on the integration of molecular data obtained by next-generation sequencing (NGS) testing.
POLYCYTHEMIA VERA
Prognostication for thrombotic events in PV
The European Collaboration on Low-Dose Aspirin in Polycythemia Vera study showed that cardiovascular event-free survival of the 1,638 patients with PV can be stratified into three groups according to age ≥65 years and a previous history of thrombosis: low-risk (neither of them), intermediate-risk (either of them), and high-risk (both of them) [1]. Based on the results of subsequent studies [2, 3], age ≥60 years (rather than ≥65 yr) and previous thrombosis have been considered as two factors predicting thrombotic events in patients with PV, consisting of the conventional risk model (Table 1) [4, 5]. Guidelines recommend that patients with low-risk PV according to the conventional risk model (age <60 yr and no prior history of thrombosis) do not need cytoreductive therapy [6, 7] unless in specific clinical subgroups, including poor tolerance to phlebotomy, symptomatic progressive splenomegaly, and persistent leukocytosis [7].
Other factors have been reported to affect the development of vascular events in patients with PV, including the presence of leukocytosis in patients aged <60 years [8-10], arterial hypertension in lower-risk patients according to the conventional risk model [11], higher intensity of phlebotomy [12], and higher JAK2 V617F allele burden [13, 14]. However, more evidence is needed regarding these variables [15], and several reports have not supported this association [16-18]. Failure to control the hematocrit increases the risk of thrombosis [19]. However, thrombocytosis did not significantly increase the risk of thrombosis. Instead, extreme thrombocytosis (>1,000×109/L) may lead to bleeding due to acquired von Willebrand syndrome [5].
Prognostication for survival in PV
Mortality in patients with MPNs results not only from thrombotic events, but also from disease progression (to MF or secondary AML) and disease-associated infections. Thus, to predict the overall survival (OS), disease-related biological risk factors should be incorporated into the thrombotic risk factors in patients with MPN. Cytogenetic abnormalities are associated with OS [20-22]. Barraco et al. [20] evaluated 196 patients with PV and showed that 19% (N=38) of them had any abnormal karyotype: presence of any abnormal karyotype and loss of Y were independently associated inferior OS, and presence of abnormal karyotype, sole abnormalities, and loss of Y were independently associated with inferior leukemia-free survival (LFS) [20]. Older age, white blood cell count >15×109/L, and use of older drugs, such as P32 and chlorambucil, have been suggested as factors affecting evolution to the blastic phase [16, 22, 23].
With the introduction of the NGS technology, interests in the effect of MPN driver mutations and other myeloid neoplasm-relevant genetic mutations on the OS of patients with MPNs increased. Tefferi et al. [24] conducted targeted deep sequencing for myeloid neoplasm-relevant 27 genes from bone marrow or blood samples from 133 patients with PV and 183 with ET. In patients with PV, ASXL1 and SRSF2 are associated with OS, and blastic transformation is influenced by mutations in SRSF2 and IDH2 [24]. The Mutation- Enhanced International Prognostic Systems for PV (MIPSS-PV) and ET (MIPSS-ET) were developed to prognosticate OS, LFS, and myelofibrosis-free survival (MFFS) in patients with ET and PV by integrating both clinical and genetic information. Among 336 patients with PV, presence of SRSF2 mutation (3 points), age >67 years (2 points; a receiver operating characteristics curve-defined age limit), leukocyte count ≥15×109/L (1 point), and history of thrombosis (1 point) were separated as independent risk factors for OS. Patients were classified into three risk groups: low-risk (0–1 points), intermediate-risk (2–3 points), and high-risk (≥4 points), and their median OS was 24 years, 13.1 years, and 3.2 years, respectively [25] (Table 1). Therefore, the validation of these results is warranted.
ESSENTIAL THROMBOCYTHEMIA
Prognostication for thrombotic events in ET
The International Prognostic Score for Essential Thrombocythemia-Thrombosis (IPSET-thrombosis) [26] is a modified version of the original IPSET [27], which aims to prognosticate OS of ET. Because IPSET was also able to predict thrombosis, an effort was made to develop a thrombosis-specific prognostic model, and IPSET thrombosis was introduced from the analyses of 891 patients with ET. IPSET thrombosis suggested that age >60 years, history of thrombosis, presence of cardiovascular risk factors, and mutated JAK2 V617F showed prognostic significance for thrombosis in multivariate analysis [26]. Further analysis of IPSET-thrombosis defined a subgroup with a very low risk of thrombosis, who were aged <60 years, had no history of thrombosis, and lacked JAK2 V617F mutation [28]. This four-group classification of patients with ET according to thrombosis risk is called the “revised IPSET-thrombosis” (Table 2). It has been validated in an independent cohort of 585 patients [29] and a retrospective cohort of 197 Japanese patients with ET [30], although prospective studies are needed for confirmation.
Previous studies showed that the incidence of thrombosis is lower in patients with CALR-mutated ET compared to those with JAK2-mutated ET [31, 32]. However, CALR mutations in patients with ET did not maintain an association with the development of thrombosis in multivariate analysis and thus were not included in the revised IPSET-thrombosis, probably because CALR mutation status tended to cluster with other lower-risk features [6, 28].
Prognostication for survival in ET
The IPSET was developed for 867 patients with ET with a median follow-up period of 6.2 years (range, 0–27) (27 yr). It includes age ≥60 years (2 points), leukocyte count ≥11×109/L (1 point), and prior thrombosis (1 point) as risk factors for inferior OS and stratified the patients into three risk categories: low-risk [0 points; median OS not reached (NR)], intermediate-risk [1–2 points; median OS, 24.5 yr; 95% confidence interval (CI), 22.3–NR], and high-risk (3–4 points; median OS, 14.7 yr; 95% CI, 11.9–18), respectively [27].
In the study, which developed MIPSS-ET (Table 2), clinical and molecular information from 451 patients with ET were analyzed [25]: age >60 years (4 points); the presence of adverse mutations SF3B1, SRSF2, TP53, and U2AF1 (2 points); male sex (1 point); and leukocyte count ≥11×109/L (1 point) were identified as independent risk factors for survival. Patients were stratified into three risk categories: low-risk (0–1 point; median, OS 34.4 yr), intermediate-risk [2–5 points; median OS 14.1 yr with a hazard ratio (HR) of 5.9 (95% CI, 4.0–8.9) compared to the low-risk group], and high-risk [≥6 points; median OS 7.9 year with an HR of 16.8 (95% CI, 10.5–27.2) compared to the low-risk group], respectively.
MYELOFIBROSIS
Prognostication for PMF
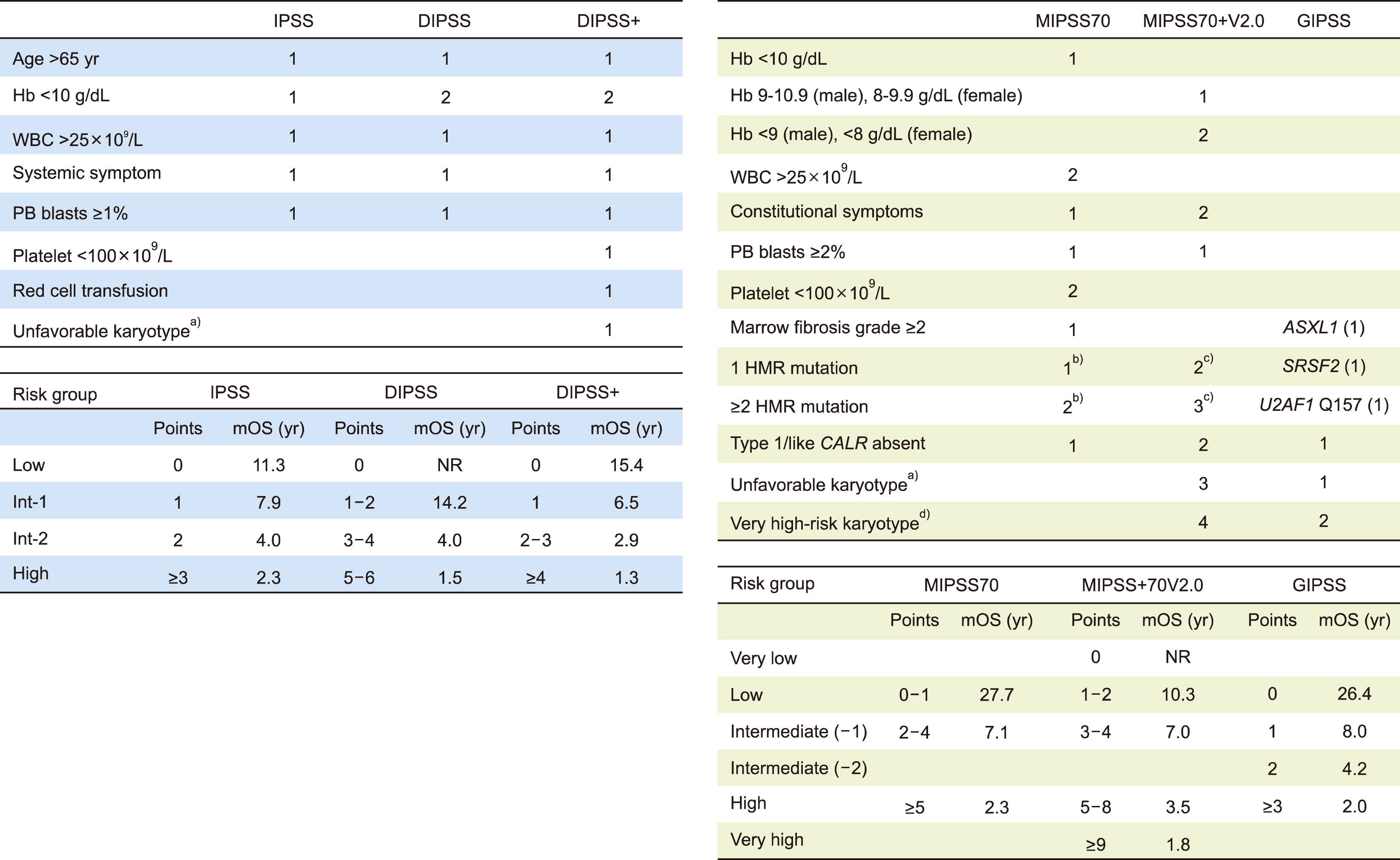
Multiple risk stratification tools have been proposed for patients with overt PMF, with or without the integration of genetic information (Fig. 1).
Mutational information not included: DIPSS and DIPSS-Plus: The International Prognostic Scoring System (IPSS) [33] is a classical prognostic scoring system for patients with overt PMF only at the time of initial diagnosis. As a dynamic risk stratification system applicable at any point over the course of overt PMF treatment is required, the impact of each adverse variable on OS during follow-up after treatment was investigated, and the DIPSS was developed [34]. The same adverse variables in the IPSS were used in the DIPSS. The only difference was that two points were assigned for a hemoglobin level of <10 g/dL, considering its stronger impact on OS [34]. The DIPSS stratifies patients into four risk groups: low-risk (0 points), intermediate-1 risk (1–2 points), intermediate-2 risk (3–4 points), and high-risk (5–6 points) with a median OS of NR, 14.2 years, 4 years, and 1.5 years, respectively [34].
Even after the suggestion of DIPSS, several DIPSS-independent risk factors have been suggested, including red blood cell (RBC) transfusion dependency, thrombocytopenia, and the presence of an unfavorable karyotype [35-37]. Therefore, it is necessary to evaluate these factors. For example, the median OS of low-risk DIPSS patients with thrombocytopenia or an unfavorable karyotype was 6.5 years compared to >15 years in the absence of these two variables [38]. Thus, DIPSS-Plus was introduced by incorporating the need for RBC transfusion, a platelet count of <100×109/L, and the presence of unfavorable karyotypes into the existing DIPSS [38]. After calculating DIPSS risk, DIPSS-Plus was calculated by adding one point each for the need for RBC transfusion, platelet count <100×109/L, and the presence of an unfavorable karyotype. The DIPSS-Plus also stratified patients into four risk groups: low-risk (0 points), intermediate-1 risk (1 point), intermediate-2 risk (2–3 points), and high-risk (≥4 points) with a median OS of 15.4, 6.5, 2.9, and 1.3 years, respectively [38]. DIPSS-Plus is particularly useful and currently recommended when karyotyping information is available; however, molecular testing is not.
Mutational information included: MIPSS70, MIPSS70-Plus, MIPSS70-Plus V2.0, and GIPSS: As genetic information acquired using NGS technology has gained popularity, mutational information-integrating risk stratification models for overt PMF have been investigated and proposed. The Mutation- Enhanced International Prognostic Score System for transplantation eligible-aged (i.e., age ≤70 yr) patients with overt PMF (MIPSS70) [39] includes hemoglobin <10 g/dL (1 point), leukocyte count >25×109/L (2 points), platelet count <100×109/L (2 points), circulating blasts ≥2% (1 point), MF-2 or higher bone marrow fibrosis grades (1 point), presence of constitutional symptoms (1 point), absence of CALR type-1 mutation (1 point), and presence of HMR mutations (ASXL1, EZH2, SRSF2, and IDH1/2; 1 point for single mutation and 2 points for ≥2 HMR mutated genes) as independent prognostic factors of inferior OS. The MIPSS70 classified the MF patients into three groups: low-risk (0–1 point), intermediate-risk (2–4 points), and high-risk (≥5 points) with a median OS of 27.7 years, 7.1 years, and 2.3 years, respectively [39]. One of the limitations of MIPSS70 is that it does not include karyotyping information. MIPSS70-Plus includes karyotypic information in the MIPSS70, and a score of 3 was assigned to an unfavorable karyotype; however, the grades of bone marrow fibrosis, leukocyte count, and platelet count were removed from the system [39]. It stratified patients with overt PMF into four groups: low-risk (0–2 points), intermediate-risk (3 points), high-risk (4–6 points), and very high-risk (VHR ≥7 points) with a median OS of 20.0 years, 6.3 years, 3.9 years, and 1.7 years, respectively.
The MIPSS70-Plus version 2.0 was introduced to 406 patients with overt PMF with fully informative cytogenetic and molecular data. It subdivided anemia into severe anemia (hemoglobin <8 g/dL in women and <9 g/dL in men) and moderate anemia (hemoglobin 8–9.9 g/dL in women and 9–10.9 g/dL in men). Additionally, it added a VHR karyotype as a separate risk factor and included U2AF1 Q157 as an HMR mutation [40]. The HR-weighted points of risk were allocated as follows: VHR karyotype (4 points), unfavorable karyotype (3 points), ≥2 HMR mutations (3 points), single HMR mutation (2 points), absence of type 1 CALR mutation (2 points), presence of constitutional symptoms (2 points), severe anemia (2 points), moderate anemia (1 point), and ≥2% circulating blasts (1 point). It classifies patients into five risk categories: very low-risk (0 points), low-risk (1–2 points), intermediate-risk (3–4 points), high-risk (5–8 points), and VHR (≥9 points) with a median OS of NR, 10.3 years, 7.0 years, 3.5 years, and 1.8 years, respectively, for patients of all ages (i.e., not limited to ≤70 yr old) [40].
The genetically inspired prognostic scoring system for PMF (GIPSS) [41] is a prognostic system for overt PMF, which comprises solely genetic information: among 641 patients with overt PMF with a complete set of both cytogenetic and mutational profile, multivariate analysis showed that VHR karyotype (2 points), unfavorable karyotype (1 point), absence of type 1 CALR mutation (1 point), and the presence of ASXL1, SRSF2, or U2AF1 Q157 mutations (1 point per each mutation) were found to predict inferior OS. The GIPSS stratifies patients with overt PMF into four risk categories: low-risk (0 points), intermediate-1 (1 point), intermediate-2 (2 points), and high-risk (≥3 points). The median 5-year OS rates were 94%, 73%, 40%, and 14%, respectively [41]. Because mutational information is acquired from the targeted sequencing of genes selected based on previous studies of prognostic relevance, GIPSS should be integrated with additional relevant genetic information that will be found in the future.
Currently, DIPSS-Plus is preferred for prognostication if a patient has cytogenetic information but lacks mutational information. If a patient with overt PMF has mutational data but cytogenetic information is not available and is 70 years old or younger, the MIPSS70 can be used. If a patient with overt PMF has both cytogenetic and mutational data, the MIPSS70-Plus version 2.0 is recommended. The MIPSS70 and MIPSS70-Plus versions were calculated online (http://www.mipss70score.it/).
Prognostication for post-ET and post-PV myelofibrosis
Although patients with post-PV or post-ET MF have different disease characteristics and natural courses from those with overt PMF [42], DIPSS and its variants have developed only in patients with overt PMF. The Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM) [43] effectively stratifies patients with post-PV or post-ET MF into four risk groups: low-risk (<11 points), intermediate-1 (≥11 and <14 points), intermediate-2 (≥14 and <16 points), and high-risk (≥16 points) according to age (0.15 points per a yr), hemoglobin <11 g/dL (2 points), circulating blasts ≥3% (2 points), CALR-unmutated genotype (2 points), platelet count <150×109/L (1 point), and presence of constitutional symptoms (1 point, Table 3). The median survival was NR, 9.3 years (95% CI, 8.1 to NR), 4.4 years (95% CI, 3.2 to 7.9), and 2.0 years (95% CI, 1.7 to 3.9), respectively [43]. The MYSEC-PM was validated in 421 post-PV or post-ET patients treated with ruxolitinib, and the prognostication of MYSEC-PM was well reproduced, whereas that of IPSS was not [44].
Treatment-specific prognostication for patients with myelofibrosis
Prognostication for patients with myelofibrosis treated with ruxolitinib: As ruxolitinib has become the standard of care for patients with a higher risk of myelofibrosis, the development of a treatment-specific prognostic model has been sought. The response to Ruxolitinib after 6 months (RR6) [45] classifies patients with myelofibrosis treated with ruxolitinib according to OS. Risk variables were as follows: receiving ruxolitinib <20 mg twice daily at all three time points (i.e., at baseline and months 3 and 6; 1 point), requirement of RBC transfusion not at baseline but at months 3 and/or 6 (1 point), achievement of <30% spleen length reduction at months 3 and 6 compared to baseline (1.5 points), and RBC transfusion requirement at all three time points (1.5 points). The median OS for patients with low (0 points), intermediate (1–2 points), and high (2.5–4 points) risk was NR, 61 months, and 33 months, respectively [45]. The investigators commented that RR6 could be a useful tool for selecting a population that needs an early shift to second-line therapy, although it needs further validation. As JAK inhibitors and other targeted agents have been investigated and introduced, a more refined treatment-specific prognostic system would contribute to improving treatment outcomes.
Prognostication for patients with myelofibrosis treated with ruxolitinib who underwent allo-HSCT: Allo-HSCT is the only curative treatment for myelofibrosis for curative intent [6]. Owing to the complexity and significant risk of non-relapse mortality after allo-HSCT, risk stratification models that can predict the outcomes of allo-HSCT in patients with myelofibrosis would be particularly useful. The myelofibrosis Transplant Scoring System (MTSS) aims to predict treatment outcomes at the time of referral for allo-HSCT in patients with overt primary or post-ET/post-PV myelofibrosis [46]. The risk variables included human leukocyte antigen-mismatched unrelated donors (2 points), non-CALR/MPL driver mutation (2 points), age ≥57 years (1 point), leukocyte count >25×109/L (1 point), thrombocytopenia (<150×109/L, 1 point), ASXL1 mutation (1 point), and the Karnofsky performance status <90% (1 point). The 5-year OS rates in the low- (1–2 points), intermediate- (3–4 points), high- (5 points), and very-high- (6–9 points) risk groups were 90%, 77%, 50%, and 34%, respectively. The HRs for death in the intermediate-, high-, and very-high-risk groups with the low-risk group (HR=1) as reference were 2.08 (95% CI, 1.1–3.8), 3.72 (95% CI, 2.0–6.9), and 6.95 (95% CI, 3.8–12.6), respectively [46]. For a better selection of patients with MF who would benefit from allo-HSCT, Passamonti [47] suggested that first, the most recent prognostic model for overt PMF, such as MIPSS70 or its variants, and post-PV/ET myelofibrosis (MYSEC-PM) should be applied to a patient with MF <70 years old to confirm the higher risk of death, which is usually defined as a median OS of <5 years, for the patient, and then the MTSS can be applied to the patient. Patients with low or intermediate MTSS risk can undergo allo-HSCT with the expectation of a lower mortality risk [47].
PROGNOSTICATION IN MPNS: FOCUSING ON MUTATION ABNORMALITIES
Table 4 summarizes the results of the analyses of certain mutations and the prognosis reported for patients with MPN. Currently, the results should be used as a component within a comprehensive clinico-hematological-genetic context rather than solely focusing on the prognostic value of individual variants/mutations [24, 25, 31, 32, 48-57].
CONCLUSION
In addition to the existing clinical understanding, a deeper understanding of the molecular aspect of the diseases enabled the development of a more accurate prognostic system in MPNs. In PV, mutations in SRSF2 are strongly associated with OS. In ET, the presence of JAK2 mutations has an impact on the development of thrombosis, and mutations in SRSF2, SF3B1, U2AF1, and TP53 adversely affect OS, whereas age and blood cell count remain strong factors affecting both vascular event-free survival and OS. In MF, adding molecular and cytogenetic information to preexisting clinical parameters significantly improved OS prediction. Thus, all cytogenetic and mutational information should be obtained at the time of the initial MF diagnosis in all patients whenever possible. If treatment-specific prognostic systems, such as RR6 and MTSS, are firmly validated, they are expected to be of great help in clinical practice, leading to improved OS.
 XML Download
XML Download