

 PDF
PDF Citation
Citation Print
Print



Introduction
Next-generation sequencing (NGS) is a widely employed sequencing platform in the era of precision medicine [1]. Conventional molecular technologies, such as Sanger sequencing and polymerase chain reaction (PCR), can cover only hotspots of limited number of genes. In contrast, NGS technology can simultaneously detect hundreds of genetic alterations [2,3]. Tumors with an identical histologic diagnosis can comprise diverse types of driver gene mutations. For example, oncogenic mutations occurring in lung adenocarcinoma include the KRAS, EGFR, ALK, ROS1, RET, MET, NTRK1–3, ERBB2, BRAF, MAP2K1, and NRG [4–6]. In addition, multiple co-occurring genetic alterations affect tumor heterogeneity and significantly contribute to its biologic behavior [7,8]. Thus, NGS is key to appropriate patient diagnosis and establishment of a treatment plan.
Currently, NGS approaches can be broadly categorized into targeted panel sequencing, whole-exome sequencing, and whole-genome sequencing [9]. Whole-exome and whole-genome sequencing approaches are widely used for research purposes; however, targeted panel sequencing is feasible for the detection of clinically relevant genetic alterations [10]. The NGS workflow involves a complex process comprising multiple steps that require optimization, familiarization, and validation [11–14]. Nonetheless, there are limited standardized validation guidelines for NGS because of the diverse types of tumor samples, available sequencing platforms, and data analysis algorithms. However, several groups have provided the validation guidelines for the NGS process [10,15]. The validation of the NGS process is critical for patient care because every step of NGS harbors potential sources of errors that can harm the patients.
In this study, we aimed to validate and demonstrate the analytical performance of ONCOaccuPanel (NGeneBio, Seoul, Korea) on the Illumina MiSeq platform. ONCOaccuPanel is a hybridization capture-based DNA panel that can detect somatic mutations and copy number alterations of 323 key cancer genes and fusions of 17 genes in solid tumors. Using numerous formalin-fixed paraffin-embedded (FFPE) samples of different tumor types, we further intended to demonstrate the clinical performance of ONCOaccuPanel. This panel is currently employed as a molecular diagnostic tool in several hospitals across Korea; thus, it is necessary to assess its clinical performance.
Materials and Methods
1. Study samples
We used 16 patient-derived FFPE samples and one reference sample (OncoSpan gDNA, cat No. HD827, Horizon Discovery, Waterbeach, UK), which comprised a wide range of mutations (single-nucleotide variants [SNVs] and insertions and deletions [Indels]) with known variant allele frequencies, for validation. The FFPE samples included samples of lung cancer (n=7), ovarian cancer (n=2), endometrial cancer (n=2), head and neck cancer (n=1), skin cancer (n=1), thyroid cancer (n=1), and malignancy of an unknown origin (n=2) (S1 Table). The tumor specimens and reference sample consisted of known genetic alterations detected by alternative molecular methods, including PCR, fluorescence in situ hybridization, and targeted NGS.
Furthermore, we investigated the analytical performance of ONCOaccuPanel in daily practice using 117 patient-derived samples (116 FFPE samples and one cytology sample). The included patient-derived samples were of 11 different tumor types—ovarian cancer (n=44), lung cancer (n=27), endometrial cancer (n=19), breast cancer (n=8), malignancy of an unknown origin (n=7), bone and soft tissue tumors (n=3), skin cancer (n=2), head and neck cancer (n=2), uterine cervix cancer (n=2), thyroid cancer (n=2), and stomach cancer (n=1). All FFPE samples displayed tumor cellularity of ≥ 10%. Two pathologists experienced in surgical pathology (Kim M and Jeong JY) reviewed the available stained slides and determined tumor cellularity. All samples were obtained from the Department of Pathology, Kyungpook National University Chilgok Hospital. S2 Table presents detailed information on the patient-derived tumor samples.
2. DNA extraction
The FFPE slices (5-μm-thick) were deparaffinized and rehydrated with xylene and alcohol solutions. We performed DNA extraction and purification using the Maxwell FFPE Plus DNA Kit (Promega, Madison, WI) according to the manufacturer’s instructions. For the cytology specimen, DNA extraction was performed using the QIAamp DSP DNA Mini Kit (Qiagen, Hilden, Germany). We measured the DNA quantity by fluorometric quantification using a Quantus Fluorometer with a QuantiFluor dsDNA system (Promega). We evaluated the DNA integrity number (DIN) using an Agilent 4200 TapeStation (Agilent Technology, Santa Clara, CA).
3. Library preparation and next-generation sequencing
For sequencing the library preparation, genomic DNA was sheared using a Qsonica sonicator (Qsonica, Newtown, CT). We measured the size of the fragmented DNA using the Agilent 4200 TapeStation and High Sensitivity D1000 ScreenTape with High Sensitivity D1000 Reagents. Library construction was performed using ONCOaccuPanel. ONCOaccuPanel DNA probes were designed for the targeted sequencing of all exons and selected introns of 225 genes and partial exons of 98 genes (a total of 323 genes). S3 Fig. presents detailed information on the gene panel. It included 3,722 coding exons, with a total target size of 1.15 Mb. We performed end repair, A-tailing, ligation, and PCR amplification according to the manufacturer’s instructions. The size of the pooled library was measured using the Agilent 4200 TapeStation and High Sensitivity D1000 ScreenTape with High Sensitivity D1000 Reagents. Library pooling, hybridization, capture, and post-capture PCR enrichment were performed according to the manufacturer’s instructions. To assess the quality and quantity of the constructed library, we used the Agilent 4200 TapeStation and High Sensitivity D1000 ScreenTape with High Sensitivity D1000 Reagents to measure the library size. The pooled libraries were sequenced on a MiSeq sequencer (Illumina, San Diego, CA).
4. Determination of coverage requirements and target region coverage
Theoretically, a true mutation with ≥ 10% frequency can be detected with 100× coverage and 98% confidence (power) (α=0.05). Thus, we were likely to capture the majority of true-positive (TP) variants with ≥ 10% frequency at 100× coverage (95% confidence interval of variant allele frequency [VAF], 4.9 to 17.6). To achieve ≥ 100× coverage for ≥ 98% of the targeted exons, the samples were sequenced to ≥ 200× mean coverage. To validate this hypothesis, we sequenced 20 clinical samples (provided by NGeneBio) using the Illumina MiSeq platform. Most genes (95.1%, 307/323) displayed > 100× coverage. The minimum coverage of the 14 essential genes (ALK, APC, BRAF, BRCA1, BRCA2, EGFR, ERBB2, IDH1, IDH2, KIT, KRAS, MET, PIK3CA, and TP53) that should be included in NGS tests in Korea is 169×. Among the 3,722 total exons, 97 displayed low coverage, i.e., < 100×. Among the 14 essential genes, exon 1 of BRAF, ERBB2, and IDH2 revealed high GC contents (77.13%, 76.52%, and 76.27%, respectively), with a low coverage, i.e., < 100×. S4 Table summarizes the coverage information of all genes, GC contents, and the list of exons with < 100× coverage.
5. Bioinformatic pipeline
Fig. 1 depicts a bioinformatics pipeline flow chart. We assessed the quality control of the FASTQ files generated by ONCOaccuPanel using FastQC (ver. 0.11.3). Subsequently, we used the Burrows-Wheeler Aligner to align reads to the human reference genome (GRCh37;hg19). Consequently, MarkDuplicates (Picard) were used to identify and tag the duplicate reads in a BAM file. We realigned the Indel mutations using the Genome Analysis Tool Kit (GATK) (ver. 4.2.6.1) IndelRealigner. The base pair quality scores were calculated using the GATK CountCovariates and TableRecalibration functions.

Fig. 1
Bioinformatics pipeline analysis process of ONCOaccuPanel: overall process (A) and SNV and Indel calling process (B). CNV, copy number variation; Indel, insertion and deletion; MAF, Mutation Annotation Format; MMR, mismatch repair; MSI, microsatellite instability; POLE, polymerase ɛ; SNV, single-nucleotide variant; SV, structural variation; VCF, Variant Call Format.
![]()
We used VarDict (ver. 1.6) for SNV and Indel calling. To reduce the false-positive calls, we filtered the following variants: (1) total read depth < 30, mutated read counts < 3, and variants with a VAF < 3% and (2) minor allele frequency > 1% in the Genome Aggregation Database, Single Nucleotide Polymorphism Database, Exome Aggregation Consortium, and Korean Reference Genome Database. All genes with blacklist variants and panel of normal were excluded from the analysis. However, well-known and clinically significant genetic alterations which are validated predictive biomarkers of drug response or have prognostic implication in a specific cancer subtype, such as EGFR exon 19 deletion or KRAS G12C mutation in lung cancer (so called whitelisted variant), were not filtered and were reviewed manually using the Integrative Genomics Viewer, even though they did not pass the filtering criteria.
For copy number variation (CNV) analysis, we used the CNVkit to estimate the copy ratios of the tumor to pooled normal samples. The copy number was measured using the following equation:
Genes with an estimated copy number ≥ 5 were reported as an amplification, whereas those with an estimated copy number ≤ 0 were reported as deletions.
We detected gene fusion using BreaKmer (ver. 0.0.2). Split (chimeric) reads with a read count < 2 were considered false positives and filtered.
The microsatellite instability–high (MSI-H) type was defined as the total number of somatic mutation (SNV/Indel) > 30 and Indel index (Indel mutation to total mutation ratio) > 20%. The MSI-H cutoff criteria was similar to that suggested in the previous study [16]. However, due to the difference in the numbers of genes included in the panel, tumor types examined, and presence of low tumor cellularity samples in clinical practice which can influence the detection of SNV/Indel, the cutoff for MSI-H was altered (SNV/Indel: from > 40 to > 30, Indel index: from 9% to 20%). Moreover, ACVR2A K43Rfs*5 mutations or oncogenic DNA polymerase epsilon catalytic subunit gene alterations, including P286R, V411L, F367C, and S279Y, were considered MSI-H types, as previously described [16].
Clinical actionability of genetic alterations was determined based on the following reference databases: Cancer Genome Interpreter (CGI), Clinical Interpretation of Variants in Cancer (CIViC), Catalogue Of Somatic Mutations In Cancer (COSMIC), Clinically relevant Variation (ClinVar), and MSK’s Precision Oncology Knowledge Base (OncoKB).
6. MSI testing
PCR using five National Cancer Institute (NCI) markers (BAT-26, BAT-25, D5S346, D17S250, and S2S123) was performed to determine the MSI status of the tumors. Representative tumor and matched normal tissues were used for MSI testing. A DNA autosequencer (ABI 3731 Genetic Analyzer, Thermo Fisher Scientific, Waltham, MA) was used to analyze the PCR products. According to the revised Bethesda Guidelines [17], tumors with at least two markers with unstable peaks were classified as MSI-H, tumors with one unstable marker were defined as microsatellite instability–low (MSI-L), and tumors with no unstable markers were designated as microsatellite stable (MSS). However, recent guidelines have demonstrated that MSI-L should be included with MSS [18]. Thus, in this study, the status of microsatellites was classified into two classes: MSI-H and MSS.
7. Mismatch repair protein immunohistochemistry
Mismatch repair (MMR) protein immunohistochemistry (IHC) was performed using primary monoclonal antibodies against MLH1 (clone 760-5091, Ventana, Tucson, AZ), MSH2 (clone 760-5093, Ventana), MSH6 (clone 790-4455, Ventana), and PMS2 (clone 288M-16, Cell Marque, Rocklin, CA) following the automated standard protocol. Mismatch repair deficient status was defined as complete absence of nuclear staining within the tumor with retained positive nuclear staining in non-neoplastic cells.
8. PCR analysis for EGFR, BRAFV600E mutation and ROS1 fusion
To detect EGFR and BRAFV600E mutations, the PANAMutyper EGFR Kit (Panagene) and the PNA Clamp BRAF Mutation Detection Kit (Panagene) were used according to the manufacturer’s instructions, respectively. Additionally, we performed real-time PCR using the ROS1 Gene Fusions Detection Kit (AmoyDx, Xiamen, China) to detect ROS1 rearrangements.
9. Statistical analyses
The positive percentage agreement (PPA) was calculated as TP/TP+false-negative, whereas the positive predictive value (PPV) was calculated as TP/TP+false-positive. We evaluated the reproducibility using Pearson correlation coefficient. All statistical analyses were performed using IBM SPSS Statistics ver. 23 (IBM Corp., Armonk, NY).
Results
1. Validation of ONCOaccuPanel
(1) Assessment of panel accuracy
We used 16 patient-derived FFPE samples and one reference material (HD827) for the accuracy test. These FFPE samples included previously confirmed genetic mutations (16 SNV, four Indels, five CNV, two structural variations [SVs], and three MSI-H types). S1 Table presents the detailed quality metrics for the FFPE samples. Briefly, all samples, except one, had an average on-target coverage of at least 250×. The average on-target rate was 65.8% (60.0%–73.0%). For the reference sample, we only included verified mutations with a known VAF (SNV 12, Inel 6) (S5 Table). For both the FFPE and reference material, the PPA and PPV were 100% for SNVs, Indels, CNV, SV, and MSI-H types (Table 1).
(2) Reproducibility and repeatability
To demonstrate the precision of ONCOaccuPanel, we performed reproducibility and repeatability tests using the reference material HD827. The SNVs and Indels were separately evaluated. The repeatability tests were performed with two replicates, each by two technicians in a single run. The reproducibility tests were performed by a single technician in three different runs. The repeatability tests showed an R2 correlation coefficient of 0.96–0.98 (Fig. 2). The reproducibility tests showed an R2 correlation coefficient of 0.97–0.98 (Fig. 3).
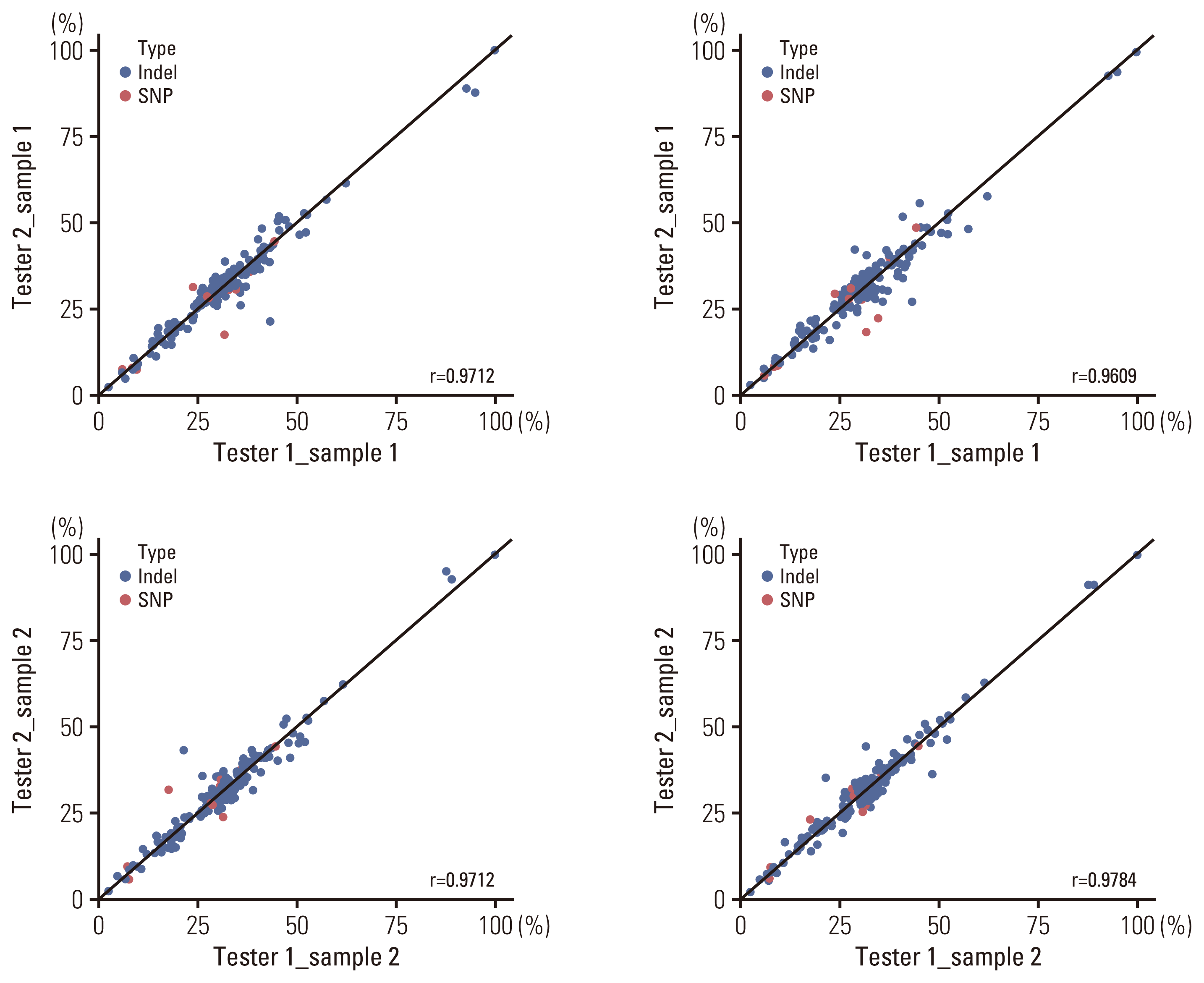
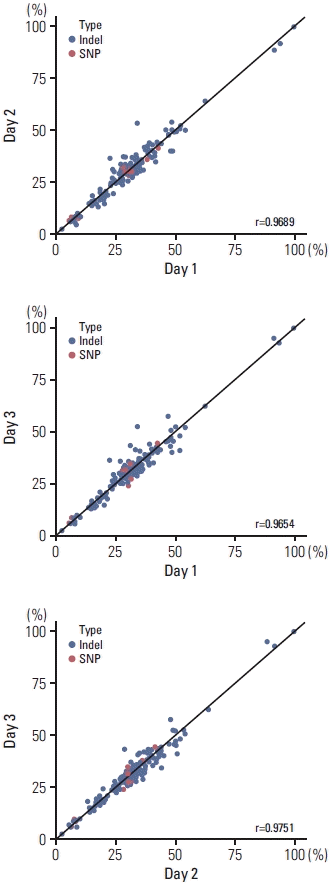
(3) Limits of detection
We evaluated the limit of detection (LOD) by sequentially diluting the reference material (1/1, 1/2, and 1/8). The LOD estimation test was performed twice in two separate runs. In both runs, all clinically relevant genes were consistently detected with a VAF of > 3% (Fig. 4). With the expected VAF of 0.5%–3%, we detected 21 of 24 (87.5%) SNVs and 11 of 12 (91.7%) Indels (Table 2).
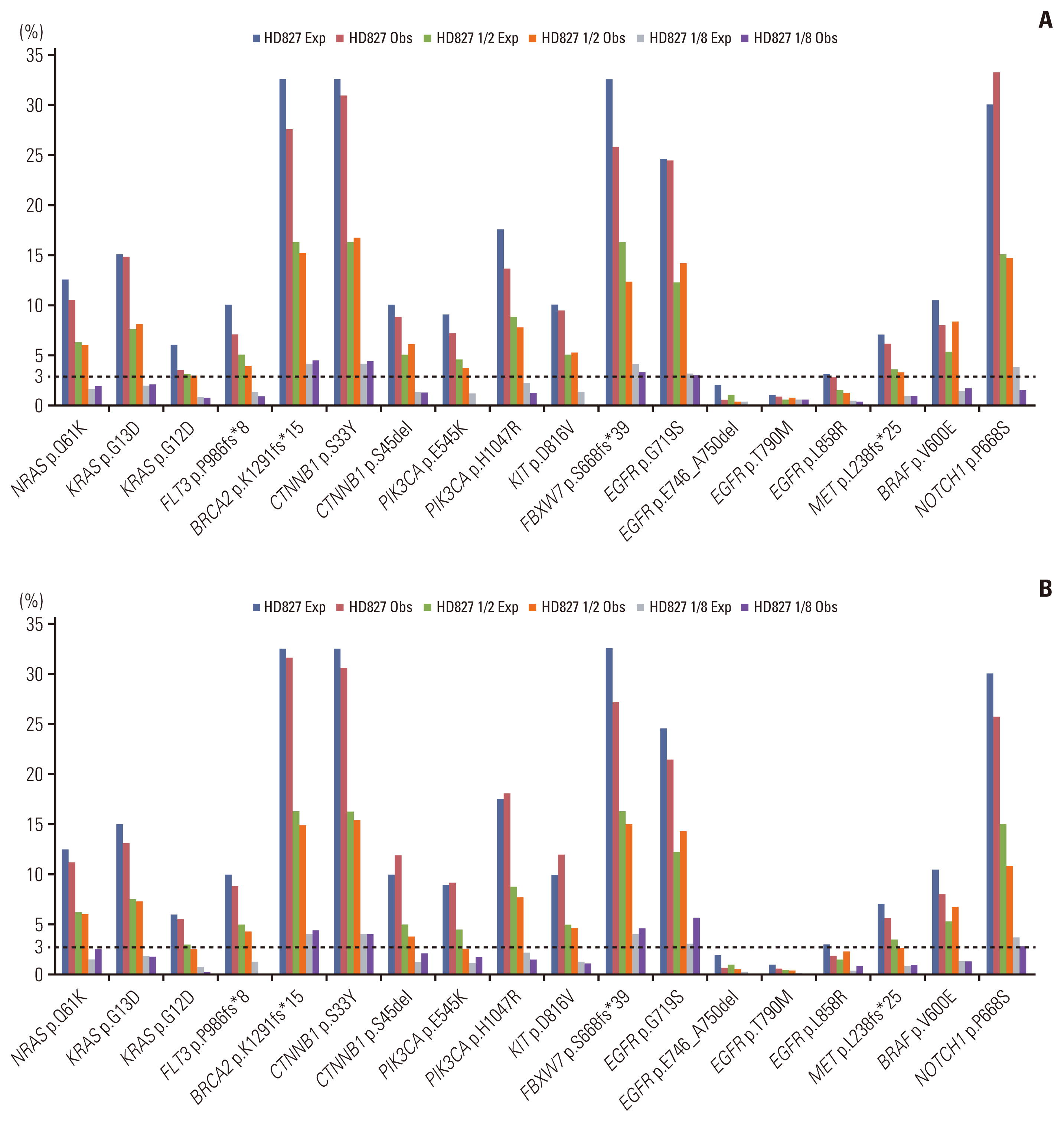
Fig. 4
Estimating the limit of detection of OncoAccupanel; first (A) and second (B) test. Percentages in the vertical axis denote the variant of allele frequency (VAF). Exp, expected VAF; Obs, observed VAF.
![]()
Table 2
Estimating the limit of detection of OncoAccupanel
![]()
2. Clinical performance
(1) Quality metrics
In the 117 clinical samples, the average total number of DNA reads was 10,500,399 (range, 4,741,992 to 15,574,808). Most samples (104/117, 88.9%) displayed an average on-target coverage of at least 250×, and all samples achieved an on-target coverage of > 100×. The average on-target rate was 64.6% (range, 48.7% to 72.5%). S2 Table presents detailed information regarding the quality metrics.
(2) Analytical performance in real-world samples
We evaluated the clinical utility of ONCOaccuPanel using clinical samples. According to the Association for Molecular Pathology/American Society of Clinical Oncology/College of American Pathologists variant categorization [19], we identified tier I (variants of strong clinical significance) or tier II (variants of potential clinical significance) variants in 115 of 117 cases (98.3%). Targetable genetic alterations, which are Food and Drug Administration–approved as predictive biomarkers of drug response, were observed in 33 of 117 cases (27.4%). Among all oncogenic mutations, the frequently altered genes were TP53 (75/117, 64.1%), ARID1A (23/117, 19.7%), PIK3CA (19/117, 16.2%), PTEN (18/117, 15.4%), KRAS (14/117, 12.0%), NF1 (10/117, 8.5%), BRCA1 (9/117, 7.7%), RB1 (7/117, 6.0%), ATM (6/117, 5.2%), PIK3R1 (6/117, 5.2%), CDKN2A (5/117, 4.3%), and other mutations with lower frequencies (Fig. 5, S2 Table). We identified oncogenic gene fusion in five cases (three RET fusions, one ROS1 fusion, and one EGFR fusion). Among all KRAS mutations, KRAS G12C mutation was identified in five cases. Among the eight breast cancer cases, PIK3CA mutations were observed in three cases. All breast cancer cases were of the triple-negative subtype. We identified complex copy number alterations in seven of eight breast cancer cases (87.5%). The MSI-H type was observed in five cases (5/117, 4.3%). All MSI-H–type cancers were uterine endometrioid carcinomas. All oncogenic mutations in BRCA1 and BRCA2 were observed in ovarian cancer. We identified gene amplification in 45 cases, and MYC amplification was the most frequently detected (11/117, 9.4%). We identified gene deletion in 17 cases, including the deletion of MTAP (6/117, 5.1%), CDKN2A (5/117, 4.3%), and CDKN2B (5/117, 4.3%). We further validated the NGS results of MSI status and other genetic alterations by PCR and IHC (S6 Table). For all five MSI-H cases, both mismatch repair protein IHC and MSI PCR testing were performed. The concordance ratio was 100% for IHC. However, one MSI-H endometrioid carcinoma with MSH6 truncating mutation (MSH6 N897Kfs*3) was MSS by PCR (concordance rate 80%, 4/5). For 112 MSS cases, PCR tests were performed in 14 cases and mismatch repair protein IHC was performed in 27 cases. The concordance ratio was 100% for both PCR and IHC. For three EGFR-mutant lung cancers, EGFR mutations were detected in two cases by real-time PCR. One EGFR wild-type by PCR test was proved to be EGFR exon 20 insertion mutation by NGS, of which the probe was not included in the commercially available kit. Furthermore, we successfully validated other genes not discussed here, which are shown in S6 Table.
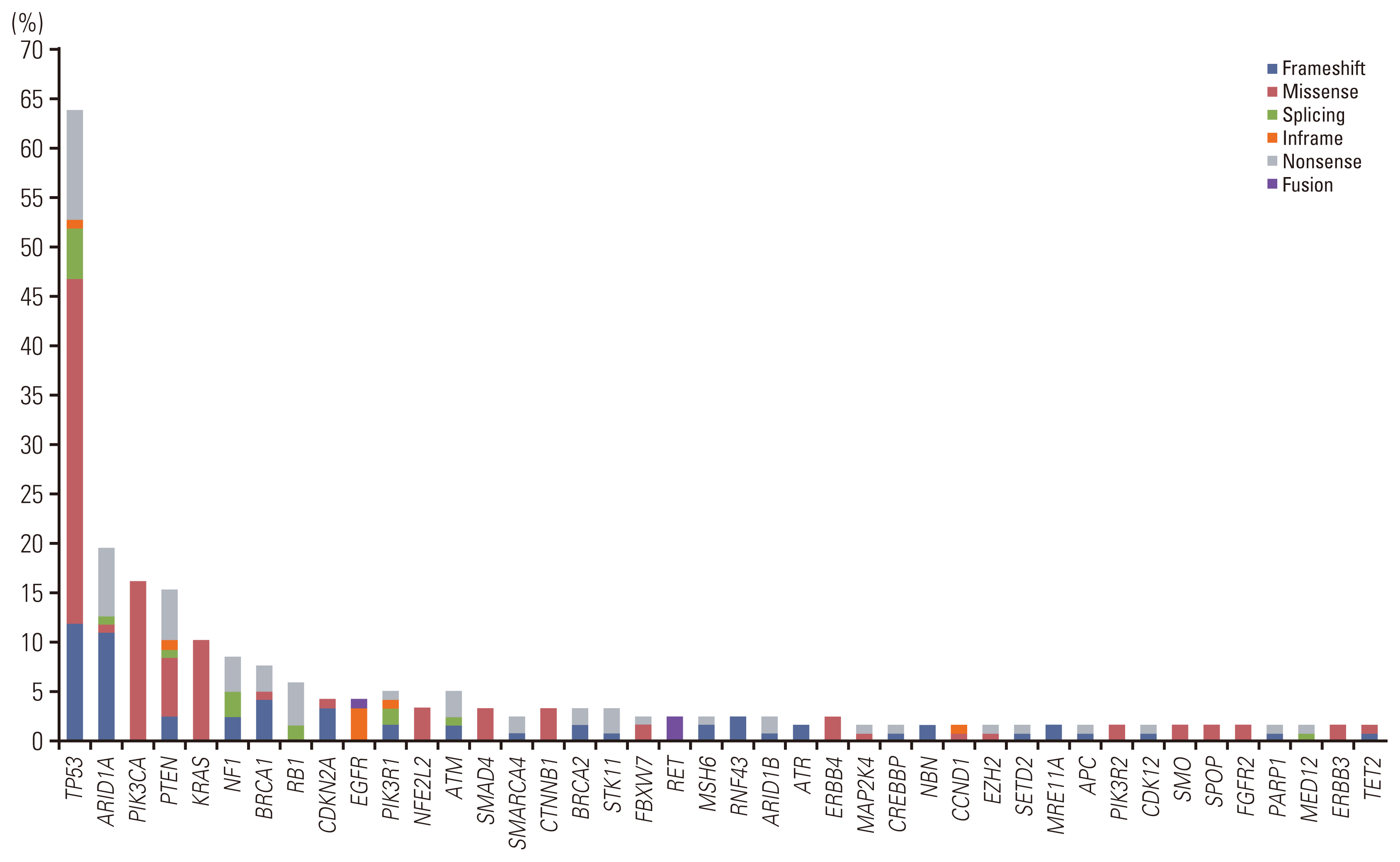
(3) Interesting cases
NGS detected TPR-ROS1 fusion in a case of lung adenocarcinoma, which was not detected by a previous ROS1 real-time PCR test. We hypothesized that it could function as an oncogenic driver owing to the retained tyrosine kinase domain of ROS1. ROS1 IHC revealed diffuse membranous positivity. Another case of lung adenocarcinoma harbored EGFR N771dup mutation, which is reportedly associated with the resistance to gefitinib and erlotinib. Considering the absence of a commercially available kit for the detection of EGFR exon 20 insertion mutations, the results of previous EGFR real-time PCR tests indicated wild-type EGFR.
Discussion
Recent advances in molecular genetics have led to the refined classification of tumors based on pathognomonic genetic alterations. The genomic profiling of tumors is essential for optimal treatment selection [20,21]. NGS can simultaneously identify the genetic alterations of hundreds of relevant genes in a single assay [22]. The NGS panel should be validated before its clinical application. In this study, we validated the analytical sensitivity, precision, and LOD of ONCOaccuPanel using patient-derived FFPE tumor samples and a reference sample, HD827. Moreover, we investigated its performance in real-world samples.
During validation, ONCOaccuPanel demonstrated excellent PPA (100.0%) and PPV (100.0%) for SNVs, Indels, CNVs, SVs, and MSI-H types. The reproducibility and repeatability tests demonstrated R2 values of 0.96–0.98. During the LOD test, we identified all clinically relevant genes with an expected VAF of > 3%, and the quality metrics for the 16 FFPE samples revealed that the target coverage for all samples was > 250× except one. This finding satisfied the recommended sequencing depth of 250× and allele frequency of 5% as the cutoff for variants [23]. One sample with < 250× on-target coverage demonstrated a DIN of < 3 (2.1).
Quality metrics using additional 117 tumor samples demonstrated an average on-target coverage of 328×. Thirteen cases revealed an on-target coverage of < 250×. The sample quality, sample input, GC content, and other factors might have contributed to the low coverage of these samples [10]. However, all FFPE and cytology samples achieved an on-target coverage > 100×, thus suggesting that we could successfully detect genetic alterations with ≥ 10% VAF. This finding is noteworthy because most solid tumor specimens available for the NGS tests include FFPE samples.
We identified a wide range of clinically relevant genes, of which TP53 was the most frequent gene. Considering the high proportion of ovarian, endometrial, and uterine cervical cancer samples, we frequently identified ARID1A, PTEN, KRAS, and PIK3CA mutations [24–27]. We identified EGFR mutations in only three of the 28 lung cancer cases. This is because most NGS performed in lung cancer comprised wild-type EGFR on previous PCR tests. Likewise, lung cancer cases were enriched with a diverse type of rare mutations, such as TPR-ROS1 fusion, EGFR-RAD51 fusion, EGFR exon 20 insertion mutation, MET exon 14 skipping mutation, ERBB2 mutation, and RET fusion. Despite limited reports of TPR-ROS1 fusion in lung cancer [28], its pathogenicity has been reported in other types of malignancies [29]. In addition, ROS1 IHC demonstrated diffuse cytoplasmic positivity, further supporting the oncogenic role in this case. All breast cancer cases included in this study were of the triple-negative subtype. Consistent with previous studies, PIK3CA was the most frequently detected oncogenic alteration, thus providing an indication for targeted therapy for breast cancer [30]. Notably, we identified complex copy number alterations in most breast cancer cases (S2 Table). This may reflect an underlying homologous recombination deficiency [31]. However, researchers should perform the loss of heterozygosity test or homologous recombination deficiency scoring test to confirm this hypothesis [32]. We also successfully validated the NGS results with conventional diagnostic methods (PCR and IHC), although relatively small number of samples were used for the validation. One MSI-H endometrioid carcinoma case (MSI-H by NGS and deficient mismatch repair by IHC) harboring MSH6 truncating mutation was MSS by MSI PCR. Although PCR testing is regarded as the gold standard to detect MSI-H tumors [33], it is well known that MSI-H tumors caused by defective MSH6 gene can be MSS by PCR analysis [34,35]. We thus concluded that MSS by MSI PCR testing in this case was a false-negative result based on the results of the NGS test (more than 30 SNVs/Indels and Indel index > 20%) and MMR protein IHC results (loss of nuclear positivity of tumor cells on MSH6 immunostaining).
This study has some limitations. We did not assess the tumor mutational burden (TMB), which can elucidate immune checkpoint inhibitor treatment [36]. ONCOaccuPanel offers TMB values; however, we were unable to compare the TMB results with those of the reference standard method. Moreover, we used relatively fewer FFPE samples and reference materials during validation. In addition, we did not include diverse types of samples; most samples were FFPE samples, and we included only one cytological specimen. However, we could demonstrate good quality metrics with good clinical sensitivity to detect the oncogenic alterations using additional 117 samples in clinical settings. Furthermore, all subsequent NGS analyses in a clinical practice setting using cytology specimens (not included in this study) demonstrated good quality metrics with an on-target coverage > 250×. In this study, gene fusions were detected by ONCOaccuPanel, DNA-based panel, including some rare types of fusions. However, the detection of gene fusions by the DNA-based panel can be influenced by several factors including the size of intronic regions with repetitive sequences. We recently experienced a lung adenocarcinoma case with CD74-ROS1 fusion that was filtered out during annotation due to the similarity of the sequence between CD74 and the intergenic region (data not included). ROS1 gene fusion was previously detected by real-time PCR. We were able to detect CD74-ROS1 fusion by manual review of the case using the Integrative Genomics Viewer. Furthermore, combined DNA and RNA sequencing can be optimal in detecting a wide range of fusions with better sensitivity, including some rare gene fusions, such as NTRK fusion [37].
In summary, we validated and investigated the clinical utility of ONCOaccuPanel, a targeted NGS panel designed to detect clinically relevant genes. This NGS panel is feasible for the detection of oncogenic alterations in clinical practice. ONCOaccuPanel is widely employed in several major hospitals in Korea; therefore, this study will supposedly contribute to patient diagnosis and treatment with the relevant molecular classification of tumors.
 XML Download
XML Download