

 PDF
PDF Citation
Citation Print
Print



Introduction
Concurrent chemoradiotherapy (CCRT) is the standard treatment option for locally advanced cervical cancer in female patients [1]. Despite its robust effectiveness, residual disease persisted in 30%–65% of patients receiving radical surgery after CCRT, and approximately 40% of these patients experienced further disease progression [2–4]. This residual disease did not affect overall survival (OS) but could be associated with decreased disease-free survival [3]. Therefore, optimal monitoring strategies need to be established for residual disease to determine progression and early intervention.
During or after CCRT, monitoring of cervical cancer has been attempted using clinical data (tumor stage, cervical cytology, human papillomavirus [HPV] status and type, and treatment modality), imaging tools (computed tomography, magnetic resonance imaging [MRI], and positron emission tomography), and tumor markers in the blood (squamous cell carcinoma antigen [SCC] and carcinoembryonic antigen [CEA]) [5,6]. However, the prognostic predictability of existing biomarkers and their ability to discriminate a residual tumor remains limited [7]. Therefore, developing a monitoring protocol with high accuracy and clinical utility remains urgent.
In cervical cancer, it has been reported that the expression of several genes is closely related to treatment outcomes [8,9]. These gene expression analyses provide physicians with additional information to monitor the presence of residual tumors after treatment. Nonetheless, invasive procedures, such as biopsy, are required, and considering the genetic diversity of each tumor, their clinical practicality remains modest. Accordingly, it is crucial to identify and develop less- or non-invasive, precision medicine-based biomarkers as potential replacements.
Combined with next-generation sequencing (NGS) techniques, liquid biopsy allows the identification of genetic information from body fluids, such as blood, less invasively than tumor tissue-based biopsies [10,11]. Furthermore, liquid biopsies can be easily obtained, thereby facilitating monitoring throughout the disease course [11]. Among liquid biopsy analytes, cell-free DNA (cfDNA) is valuable for post-treatment monitoring of various carcinomas [12,13]. Quantifying HPV DNA and tumor-specific mutations, which remain the major underlying cause of cervical cancer, in plasma cfDNA could drastically improve monitoring accuracy after treatment [14–18].
Herein, we aimed to investigate a liquid biopsy-based monitoring method to identify residual cervical cancer after radical radiotherapy (RT). Furthermore, we compared the accuracy and efficacy of the developed method with those presented by contemporary surveillance strategies using MRI and tumor markers in patients with cervical cancer undergoing RT.
Materials and Methods
1. Prospective cohort and samples
Inclusion criteria were as follows: (1) patients with pathologically proven uterine cervical cancer; (2) patients aged ≥ 20 years, with an Eastern Cooperative Oncology Group performance status of 0–2; (3) patients who had completed planned radical RT; and (4) patients without distant metastasis (DM). Patients were excluded if they had current or previous malignancies within 2 years of enrollment. Twenty-eight patients (CX-001~CX-028) were recruited, but three subsequently withdrew consent. The median follow-up duration was 25.4 months (range, 1.9 to 34.1 months).
Peripheral blood was drawn from each patient, and pelvic MRI was performed before RT (visit 1), during RT (especially before brachytherapy, visit 2), and three months after RT (visit 3). In 14 patients, cfDNA was obtained three times (i.e., visits 1, 2, and 3). Eleven patients were referred for brachytherapy from other hospitals; therefore, they provided blood samples at visits 2 and 3, respectively. Finally, we analyzed 64 plasma cfDNA samples from 25 participants. We also evaluated the known tumor markers, SCC (for squamous cell carcinoma) and CEA (for adenocarcinoma), to compare the therapeutic monitoring values of cfDNA.
2. Plasma cfDNA and genomic DNA library preparation
Library preparation and sequencing were performed by Celemics (Seoul, Korea). cfDNA and genomic DNA (gDNA) were extracted from the blood samples for library preparation. gDNA was fragmented to approximately 250 bp using the Bioruptor Pico Sonication System (Diagenode, Belgium). cfDNA and fragmented gDNA were processed for Illumina sequencing (San Diego, CA) using the following steps: end repair, dA-tailing, adapter ligation, and pre-polymerase chain reaction (PCR) for the indexed NGS library. The prepared cfDNA or gDNA library was hybridized with capture probes to capture target regions using the Celemics Target Enrichment Kit (Celemics). Capture probes were designed and chemically synthesized to hybridize the target region (67 oncogenes: ABL1, AKKT1, ALK, APC, ARID1A, ATM, BRAF, CASP8, CDH1, CDKN2A, CSF1R, CTNNB1, DPP4, EDN3, EGFR, EP300, ERBB2, ERBB3, ERBB4, FBXW7, FGF14, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, IGF1R, JAK2, JAK3, KDR, KIT, KRAS, MAPK1, MET, MLH1, MPL, NFE2L2, NOTCH1, NPM1, NRAS, PARP1, PDGFRA, PIK3CA, PRKDC, PTEN, PTPN11, RB1, RET, SHKBP1, SLC2A1, SMAD4, SMARCB1, SMO, SRC, STK11, TAC1, TGFBR2, TP53, VHL, WNT16, XRCC5, and XRCC6; HPV types 16 and 18). For sample enrichment, the captured regions were further amplified by post-PCR. The target-captured libraries were then sequenced on an Illumina NextSeq550 instrument using a 2×150 bp read layout.
3. Human data pre-processing
After sequencing the libraries, Trimmomatic (ver. 0.33) and BWA (ver. 0.7.10) were used for sequence-read filtering and alignment to the human reference genome. UCSC hg19 was used as the reference. Genome analysis tool kit (GATK) tools were used to pre-process the read data. PCR duplication removal, INDEL realignment, base quality score recalibration, variant calling, and variant filtration were sequentially performed using GATK. We conducted these steps for 64 cfDNA and 64 gDNA samples collected from 25 patients at three-time points and detected 32,045 variants. To identify cfDNA variants from tumors, we removed variants in both cfDNA and gDNA for each patient and at each time point. Using these steps, we finally identified 21,866 variants used in the statistical analysis. To assess the effects of each variant allele on the respective transcript, we used the ENSEMBL IMPACT rating system and selected HIGH-grade variants.
4. HPV data pre-processing
Given that HPV-originated and human DNA were mixed in blood, we divided cfDNA sequence reads for HPV quantification. Therefore, we used cfDNA reads that were not aligned to the human reference genome and aligned them to the HPV reference genome (types 16 and 18) using HPViewer. The HPV cfDNA ratio (HPV ratio) was calculated as follows:
5. Mutated gene list from cBioPortal
We used the mutated gene list from The Cancer Genome Atlas (TCGA) for cervical squamous cell carcinoma and endocervical adenocarcinoma, downloaded from cBioPortal (https://www.cbioportal.org/).
6. Statistical analysis
Progression-free survival (PFS) was defined as the time from enrollment to the date of locoregional failure or DM. According to the International Federation of Gynecology and Obstetrics (FIGO) stage and initial HPV ratio, Kaplan-Meier curves of PFS were generated and compared using the log-rank test.
The patients were divided into two groups according to their disease status after treatment: the DM group, who experienced DM until their last follow-up, and the no evidence of disease (NED) group, which was tumor free. For Fisher exact test between patient groups (DM and NED), we examined the variant allele frequency (VAF) of variants in each patient. We compared the proportion of increase or decrease for each VAF from samples collected during RT (visit 2) and those after RT (visit 3) between the DM and NED groups. Fisher exact test was used to determine statistical significance. The significance judgment cutoff was 0.100 owing to the small number of patients with DM.
All statistical analyses were performed using R statistical software ver. 4.1.2 (https://www.r-project.org/).
Results
1. Baseline characteristics
Table 1 summarizes the clinical and histopathological data of 25 patients with cervical cancer. The median age of the patients at diagnosis was 52 years. Of these, 20 patients (80%) had squamous cell carcinoma, and most (n=21, 84%) had stage III disease. Tissue samples of 13 patients (52%) exhibited HPV positivity. Except for one patient, all participants received concurrent RT and chemotherapy. All patients were administered external beam RT, followed by brachytherapy. After radical RT, all patients achieved complete remission; however, three patients experienced DM without locoregional recurrence. All 25 patients were alive at the last follow-up.
2. Mutational landscape of participants
In total, 21,866 cfDNA variants were detected in 64 samples from 25 patients, with each sample presenting 342 variants on average (S1 Table). The three samples with the highest mutational burden were from CX-024 at visit 1 (n=1,243), CX-021 at visit 3 (n=1,163), and CX-028 at visit 1 (n=1,081). In contrast, the CX-001, CX-003, and CX-004 samples from visit 1 exhibited less than 100 variants. On average, 326 variants were present in each gene. ARID1A (n=1,198, 5.5%) exhibited the highest number of variants, followed by ERBB2 (n=901, 4.1%) and PRKDC (n=850, 3.9%). Among the 67 oncogenes, TAC1 was the most highly preserved in our study population (n=28, 0.1%). In TCGA data (S2 Table), PIK3CA had the largest proportion of mutated genes (27.3% of all samples) among 796 cancer genes. However, our analysis revealed that alterations in PIK3CA accounted for only 0.8% of variants. The missense variant (n=6,453, 26.3%) was the most common variant type, followed by intron variants (n=6,307, 25.7%) and frameshift variants (n=5,125, 20.9%). The total number of HIGH-grade variants was 5,807, with a frameshift variant of 88.3% (Fig. 1A, S3 Table). In particular, the top listed genes in the oncoplot exhibited ≥ 2 different alterations in one patient. We also reported the dynamic variant changes in genes in each patient during follow-up, as shown in Fig. 1B.
3. Therapeutic monitoring and prognostic values of HPV ratio
To quantify the HPV cfDNA in each sample, we determined the HPV-originated sequence reads and their ratios when compared with all aligned sequences of human and HPV reference genomes. HPV cfDNA was detected in 14 patients (Fig. 2); interestingly, conversion of HPV types in cfDNA was identified in two patients: CX-005, HPV 18 (0.00120%)–HPV 18 (0.00017%)–HPV 16 (0.00001%); and CX-016, no sample–HPV 16 (0.00002%), HPV 18 (0.00026%)–HPV 18 (0.00001%). We depicted changes in the HPV ratio from visits 1 to 3 along with tumor markers and size simultaneously, as shown in Fig. 2. The tumor markers and HPV ratio tended to decrease as the tumor shrank; however, the effect was not proportional (S4 Fig.).
The HPV ratio of CX-011 peaked at visit 2 and subsequently decreased. In contrast, CX-004 and CX-027 presented an increased HPV ratio obtained from the last blood sample, and both experienced DM. Blood samples were collected after and before DM for these patients (CX-004 and CX-027). In terms of tumor markers, a slight increase was frequently observed, and a dramatic increase in tumor markers was detected in two patients (CX-020 and CX-028). However, only CX-028 exhibited peritoneal metastasis during treatment.
We summarized the typical temporal changes between visits 2 and 3, including tumor size, tumor markers, and HPV ratio, emphasizing better sensitivity of the HPV ratio (Table 2). In two patients from the DM group, the HPV ratio increased more than the tumor markers, i.e., the HPV ratio increased by more than 900%, but the tumor marker showed an increase of only up to 100%. The NED group demonstrated a discrepancy between the tumor markers and HPV ratio. Although not a major change, tumor markers were numerically increased in the NED group, whereas all HPV ratios were notably decreased.
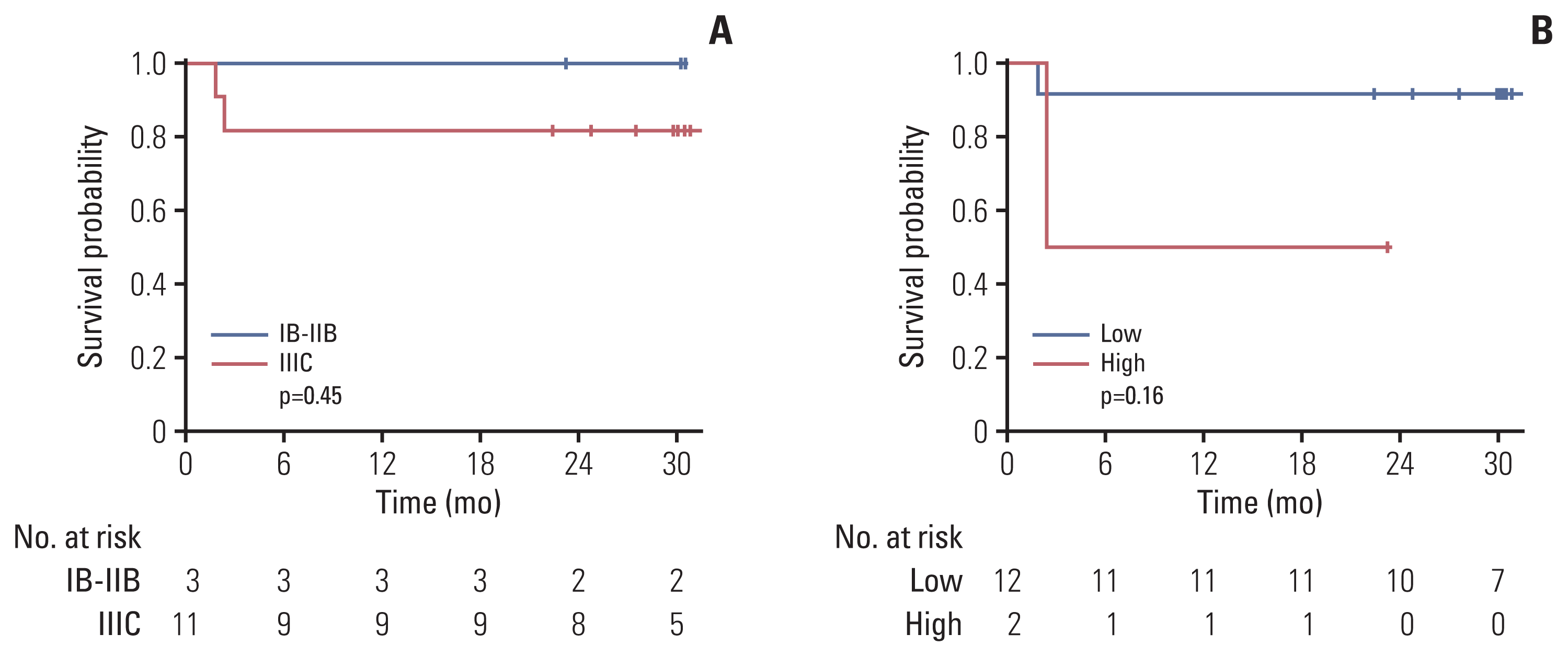
To assess the prognostic value of the initial HPV ratio (n=14), we calculated and compared the PFS according to the FIGO stage and initial HPV ratio (based on the average value of the initial HPV ratio of 0.13607%) (Fig. 3). Although no statistical significance was detected in either analysis, patients with a high initial HPV ratio seemed to exhibit a poor prognosis (p=0.16). Furthermore, fewer patients were classified in the high initial HPV ratio group (n=2) than in the FIGO stage IIIC group (n=11).
4. Time-related changes in VAF between the DM and NED groups
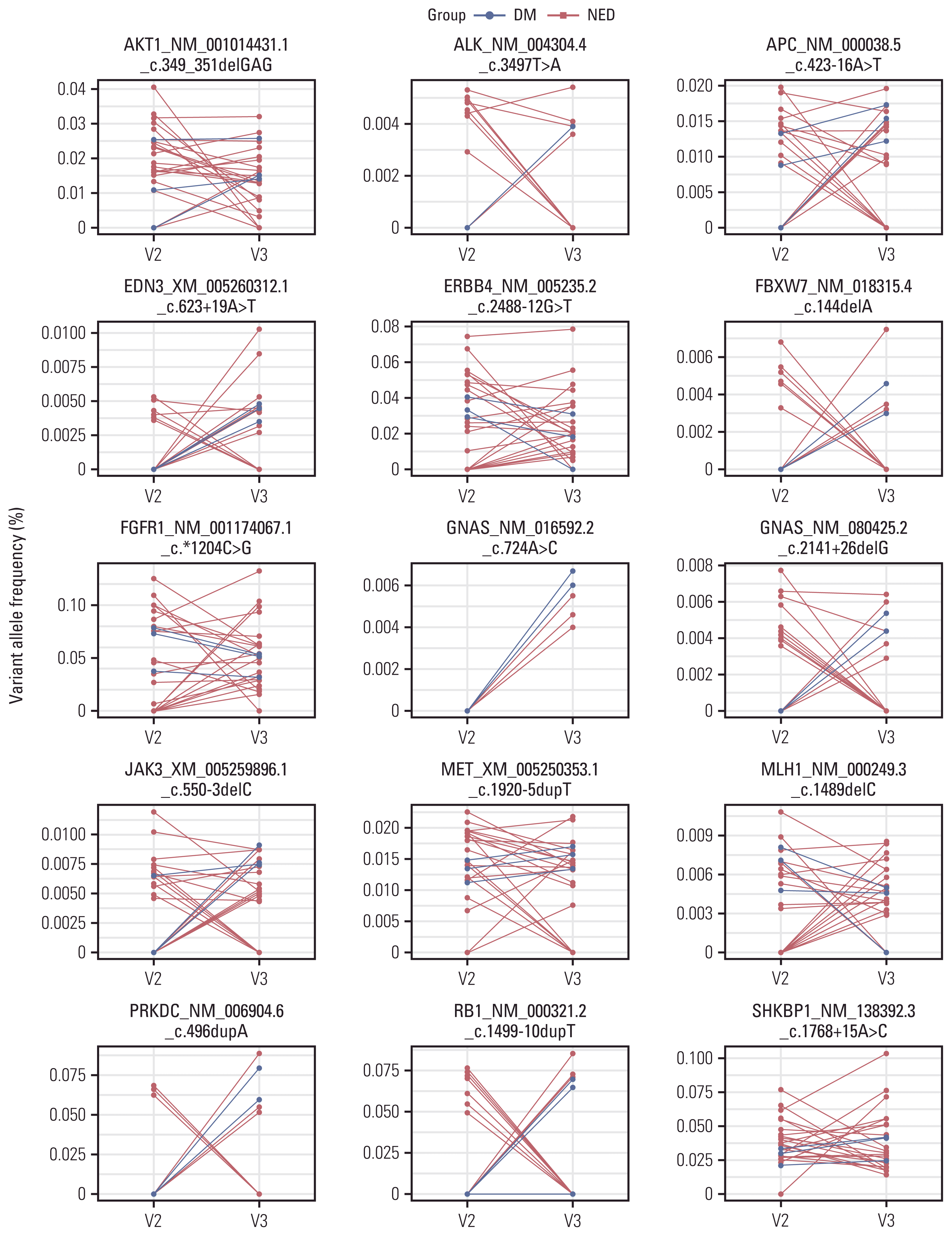
We demonstrated that VAF was found only in cfDNA but not in gDNA. Considering the relationship between oncogenes and tumor cells, variants found only in cfDNA could indicate their potential tumor cell origin. We performed a statistical analysis to identify DM group-specific variants. A detailed description of the Fisher exact test is provided in the Materials and Methods section. Fifteen variants were identified (Fig. 4). Among these, the VAF of 12 gene variants increased after treatment in the DM group. However, no consistent trend in VAF was noted in the NED group.
Discussion
In the present study, we investigated cfDNA to facilitate genetic profiling of patients with cervical cancer using amplification-based NGS, targeting 67 oncogenes and HPV type 16/18. We observed comprehensive and dynamic cfDNA alterations in these patients. Their distribution markedly differed from that in TCGA database. Herein, we revealed that the HPV ratio has the potential to be a novel biomarker with high sensitivity and specificity. Our analysis did not prove the practicality of VAF in oncogenes, possibly owing to a few treatment failures.
Sequencing samples were derived from the plasma of each donor. We analyzed variants that were only identified in cfDNA and excluded those found in gDNA, given that we regarded gDNA variants as unique mutations in individuals that are not found in all tumors. For all samples, 21,866 cfDNA variants were recognized and summarized in chronological order. We documented an actively changing tumor population in each patient according to the time point. These allele alterations might be affected by chemotherapy and RT [18]. Specific tumor cells harboring treatment-sensitive mutations were eradicated, although even new mutations may also arise. This complicated tumor environment can be utilized to monitor the disease course [18].
ARID1A (AT-rich interactive domain 1A) was the most frequently altered gene in our samples, which differed from the results based on TCGA analysis and previously reported Korean data [18]. ARID1A encodes BAF250a, a nuclear protein, and its location is susceptible to deletion [19,20]. In our analysis, all patients exhibited a frameshift variant in the ARID1A gene. ARID1A has been known as a tumor suppressor gene in several cancers, including cervical cancer [20–23]. Notably, loss of BAF250a expression can be associated with poor OS [20]. Examining this aspect might reveal valuable prognostic information. Moreover, ARID1A and ARID1A-related pathways might be innovative targets for treating cervical and other cancers.
Findings from seven representative patients revealed that the HPV ratio might substitute current tumor markers, as both are determined from blood collected by single-needle punctures. We used the HPV ratio to adjust the variant total sequencing read counts for each NGS. The HPV ratio responded more sensitively to intrinsic tumor burden and emphasized that primary tumor regression in imaging modalities did not always ensure successful treatment completion [24]. The increased HPV ratio might indicate the existence of treatment-resistant tumor cells. Although further validation is necessary, a high HPV ratio before treatment may be associated with an inferior prognosis. Although the results were drawn from a small number of patients, the HPV ratio might have a higher accuracy in risk stratification than that of the FIGO stage: among 11 patients with FIGO stage IIIC, only two patients were assigned to the high initial HPV ratio group.
We hypothesized that increased cfDNA variants in a patient after radical RT might reflect treatment resistance and imply locoregional recurrence and/or DM. Previous studies have demonstrated that specific gene mutations in cfDNA are associated with survival outcomes [16–18]. PIK3CA mutation is one of the most typical findings in such studies [16,17]; however, our results did not include PIK3CA variants (Fig. 4). Regarding the proportion of VAF-increased patients, 11 genes with 12 variants were identified in the DM group compared with those in the NED group: AKT1, ALK, APC, EDN3, FBXW7, GNAS, JAK3, MET, PRKDC, RB1, and SHKBP1. Given that only three patients presented with treatment failure (DM group), further validation is needed to establish whether the increment in these variants was statistically significant. Furthermore, this analysis highlights the importance of optimal blood sampling intervals after treatment. Currently, it remains unclear whether the increased specific VAF can be attributed to an increase in a specific tumor cell population (i.e., treatment-resistant) or an increase in treatment-mediated lysis (i.e., treatment-sensitive).
This study had some notable limitations. This prospective cohort study aimed to explore the potential clinical application of cfDNA using a liquid biopsy method. Therefore, the study population was small, and a limited number of events, such as DM, were documented. This complicated the determination of statistical significance. All samples were obtained from patients with cervical cancer, and there were no comparable control data from healthy participants. Furthermore, we did not obtain sequencing data from white blood cells or tumor samples. These two factors may induce excessive noise during variant calling. Although imaging for residual disease after treatment completion can indicate complete remission, pathological confirmation of residual disease was not performed in the present study. More long-term follow-ups and additional time points for blood sampling during follow-up are required to validate the therapeutic monitoring and prognostic role of the HPV ratio or VAF in oncogenes. Despite these drawbacks, the main advantage of the current study was that our participants were considerably homogenous in terms of FIGO stage and treatment modalities.
In conclusion, we successfully established a liquid biopsy-based monitoring platform combined with high-throughput NGS for patients with cervical cancer after radical RT. Among the dynamic genomic changes in cfDNA in response to treatment, we confirmed that HPV ratio rather than HPV read counts could be a powerful tool for surveilling and predicting treatment outcomes. Once validated, these approaches could improve daily clinical practice. Regrettably, we failed to obtain meaningful insights into the VAF changes in oncogenes, and further large-scale studies are warranted.


 XML Download
XML Download