

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Introduction of biological therapies in form of monoclonal antibodies or soluble cytokine receptors have led to dramatic improvements in the management of several debilitating immune-mediated inflammatory diseases including rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriasis, psoriatic arthritis, CD and UC.123 Especially, the use of tumor necrosis factor alpha (TNF-α) antagonists, such as adalimumab, certolizumab, etanercept, golimumab, and infliximab especially applied early in the disease course have been shown to induce clinical remission and ward off structural damage by modifying disease progression, thus, resulting in reduced the need for corticosteroid treatment, hospitalization and surgery and improved quality of life of many patients. 4 The incidence of some chronic IBD such as CD and UC is rising, which may increase the number of patients who are appropriate candidates for biological therapies.56
The biological agents are large and very complicated molecules, approximately 1,000 times larger than chemical drugs, which are produced by living cell cultures, thus requiring huge investment. The long duration of development and high production costs are cited as the main contributors to the high price of biological agents, therefore, prolonged use of these agents may be very expensive, placing a serious burden on National Healthcare Systems.7 As the patents for several TNF-α antagonists used in inflammatory diseases have or will soon expire in many countries around the world, the development of biosimilars has become another way to improve patient's outcomes and potentially lower healthcare costs. In this review, I aim to explore some of the most important aspects of the biosimilars focusing on their role in IBD.
WHAT IS BIOSIMILAR?
A biosimilar is a protein-based medical product developed using recombinant DNA technology that has a molecular structure and biological properties highly similar to the innovator product that has been approved by drug related authorities, such as the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA). The FDA definition of a biosimilar makes this clear; a biosimilar is a biological product that is highly similar to the reference product not withstanding minor differences in clinically inactive components and that there are no clinically meaningful differences between the biological product and the innovator product in terms of the safety, purity, and potency.8 The manufacturing of biosimilar is more complex than the production of generic chemical drugs. It requires several steps, including the determination and growth of a vector, the host cell expression system, the cell expansion procedure, the protein recovery mechanism, the purification process and the formulation of the therapeutic protein into a drug.9 Even after patent expiration, manufacturing processes do not have to be disclosed, so there are likely to be appreciable differences in the manufacturing processes of biosimilars and their innovator product. Biosimilars are not generic products, so it is therefore likely that these differences in the manufacturing process will lead to subtle differences between them.
Drug related authorities such as EMA, FDA, and Health Canada hold their own guidance on requirements for demonstration of the similar nature of two biological products in terms of safety and efficacy. The EMA developed a specific legal pathway for sanction of biosimilar, in which it is recognized that biosimilar may not be identical, but must be similar to the original EU-approved innovator products in terms of efficacy, safety, and quality.10 The Biologics Price Competition and Innovation Act11 designates an abbreviated licensing pathway for biosimilar in the USA, but the most stringent criteria for the approval of biosimilar are those of Health Canada. To date, a total of 20 biosimilars have been evaluated and 15 authorized in the EU. Based on the EMA and the World Health Organization (WHO) guidelines, several countries including Australia, Canada, China, India, Japan, Korea, and others have established or are establishing their own regulatory pathways for biosimilar development.
Biological agents, including adalimumab and infliximab, are seven of the 10 top grossing drugs in the world.8 For instance, the sales of infliximab in 2013 were 4 billion USD in USA and 2.2 billion USD in EU.12 The cost savings by using biosimilars are anticipated to be 25%–70% compared to their innovator product and this can make it easier to use biological agents for patients in need. Another possible benefit is the development of biobetter molecules which target the same validated epitope as the innovator product, but have been genetically modified to improve their pharmacokinetic properties, for example, optimization of glycosylation profiles to enhance effector functions or exchanging of Fc domains to increase serum half-life or antibody-dependent cellular cytotoxicity.13
WHAT ARE THE REGULATORY REQUIREMENTS?
It is common for registered biological agents to undergo manufacturing process improvements and changes during the life cycle of the drug after approval. Even minor adjustments in manufacturing processes can change biological functions, efficacy or safety profile of the same biological agent. When such a change takes place, the manufacturer should carry out a comparability exercise to prove that the change does not adversely affect the identity, purity, or potency of the agent. For example, during the manufacturing process of recombinant erythropoietin product, a replacement of human serum albumin as a stabilizer with the synthetic detergent polypsorbate 80 and glycine, resulted in an immune response against membrane-bound erythroblasts in the bone marrow. This minor change in the manufacturing process induced the formation of autoantibodies to both endogenous and exogenous erythropoietin, increasing the incidence of pure red cell aplasia, a rare but fatal disease.14 Neutralizing antibodies to erythropoietin were also produced by the biosimilar HX575 because of tungsten contamination during the making of the syringes which caused protein denaturation and aggregation of HX batches.15 Virtually all monoclonal antibodies are immunogenic and can induce antidrug antibody formation. The development of antidrug antibody may lead to accelerated drug clearance, reduced drug efficacy and higher risk of infusion reactions. Therefore, immunogenicity of monoclonal antibodies is a major safety concern for the biosimilars not only by many clinicians but also the regulatory authorities.
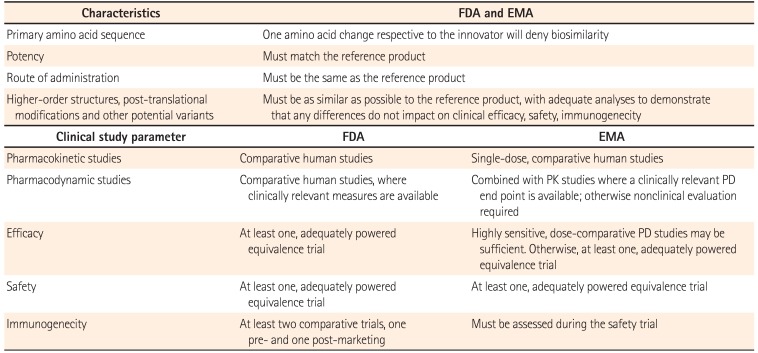
The principles conducting the comparability exercise required for biosimilar development are similar to those for innovator product changed manufacturing process, however, are more extensive and require at least one clinical study. Therefore, the key question for biosimilars is not whether differences exist compared with the innovator product, whether such differences are clinically significant. The EMA and FDA announced specific requirements for the comparability exercise of biosimlar development (Table 1).81016 For example, both the EMA and the FDA require that the primary amino acid sequence, potency, dose, and route of administration of the biosimilar must be the same as the innovator product. Higher-order structures, post-translational modifications and other potential variants have to be as similar as possible to the innovator product, with adequate analyses to prove that any difference do not make an influence on clinical efficacy, safety, or immunogenicity. Both the EMA and the FDA require one or more adequately powered equivalence trials to demonstrate similar efficacy, safety and immunogenicity for the biosimilar and innovator product.
EXTRAPOLATION ISSUES
The biosimilar to infliximab, CT-P13 has been approved recently in several countries based on a single equivalence trial conducted in patients with RA,17 supplemented by a pharmacokinetic study on AS.18 In addition to approving the CT-P13 for RA and AS, the EMA and Korean FDA allowed full label extrapolation including CD, UC, psoriasis and psoriatic arthritis although this agent was not tested in these indicated patients, other regulatory agencies allowed partial extrapolation.15 Health Canada approved the CT-P13 for all indications except CD and UC based on the differences including FcγRIIIa receptor binding, the level of afucosylation and some antibody-dependent cell-medicated cytotoxicity (ADCC) issue.19 However, the initial differences detected on ADCC were only seen in vitro using target cells that were engineered to overexpress membrane TNF and using enriched natural killer cells from CD patients with the high affinity genotypes of the FcR. When ADCC activity was tested using more physiologic effector cells such as whole blood or isolated peripheral blood mononuclear cells, the difference in fucosylation for CT-P13 and the innovator drug did not impact ADCC,20 questioning the clinical significance of the observed differences in FcγRIIa binding. Another important consideration for bioequivalence testing is to select a patient population and clinical end point most sensitive to detect clinically significant differences in efficacy and safety. If the difference in efficacy between a test drug and placebo is small, it is difficult to prove a difference between them even if there is. If a biologic agent is shown to be biosimilar in the highest placebo-adjusted efficacy model (such as those of the highest response rate and lowest placebo effect), it may be approved for use across all indications. Of the six indications of infliximab, the greatest placebo-adjusted response was found in plaque psoriasis, followed by psoriatic arthritis and CD. In contrast, RA revealed the smallest placebo-adjusted response, in other words, RA is likely to be a less sensitive clinical model to demonstrate a potential difference in efficacy between CT-P13 and infliximab.21 The development of antidrug antibodies or Immunogenicity is the primary safety concern for biological agents, therefore, the regulatory agencies require that the immunogenicity profile of biosimilars be sufficiently characterized. The immunogenicity profile should be studied in the patient population with the highest risk of an immune responses.8 The highest incidence of immunogenicity was detected in CD,22 up to 61%, whereas, it was lowest in RA, less than 10%23 which was even further suppressed by the concomitant use of immunosuppressant.24
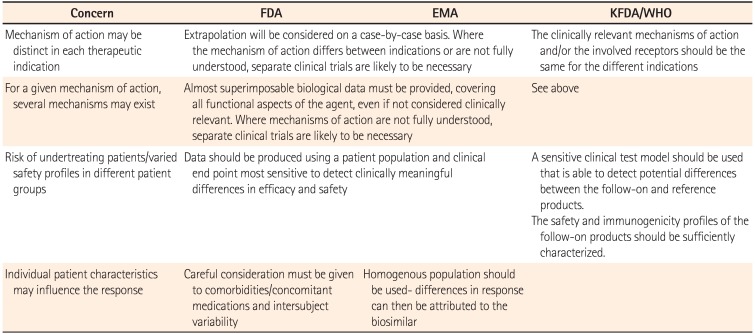
Extrapolation into other indications is essential to keep the cost of biosimilars competitive. The decision to extrapolate should be based primarily on the demonstration of similarity through extensive comparability studies that compare the physicochemical attributes and the biological activity between the biosimilar and innovator product provided by the regulatory guidelines. If all indications have to be studied, time-consuming prior authorization would minimize the cost reducing effect of biosimilar. The EMA, the FDA and the Korean FDA provide the regulatory guidelines regarding extrapolation of clinical data for biosimilar (Table 2).21
INTERCHANGEABILITY AND AUTOMATIC SUBSTITUTION
Interchangeability between the innovator product and its biosimilar remains an important subject for discussion. As defined by the FDA, interchangeability indicates that biosimilar may be expected to demonstrate the same clinical result as the innovator product in any given patient and if biosimilar is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between the use of biosimilar and the innovator product is not greater than the risk of using the innovator product without such alteration or switch.7 Thus, interchangeability represents a higher standard than demonstrating biosimilarity to the innovator product. At the present time, the FDA has not yet provided guidance on the method of studying needed to enable a determination of interchangeability.
The American Academy of Dermatology has guided its recommendations for the process of substitution of a biosimilar for a innovator biologic (Table 3).24 Most clinicians argue against the interchangeability of biosimilars.
In a head-to-head comparison cross-over study, switching from epoetin-α to the epoetin biosimilar SB309 resulted in, approximately, a 10%–15% increase in the dose required and transiently decreased the hemoglobin concentration by 5%.25 Switching back from SB309 to epoetin-α reduced the dose required by almost 10% and increased hemoglobin levels by 10%. The proponents of interchangeability argue that changes, to date, in the manufacturing process did not preclude full interchangeability.26 Indeed, most clinicians were not aware of these changes in manufacturing. The EMA has recently designated a biosimilar as automatically substitutable, although each country will follow its own national guidelines.
CLINICAL STUDIES IN IBD
Currently, there are no prospective randomized non-inferiority trials evaluating the clinical efficacy and safety, as well as interchangeability of a biosimilar in IBD patients. We performed a retrospective multicenter study evaluating the clinical efficacy and safety of CT-P13 in 32 anti-TNF naïve CD patients and 42 anti-TNF naïve UC patients.27 In anti-TNF naïve CD patients, clinical response rates at week 8, 30 and 54 were 90.6%, 95.5% and 87.5%, respectively, and clinical remission rates at week 8, 30 and 54 were 84.4%, 77.3% and 75.0%, respectively. In anti-TNF naïve UC patients, clinical response rates at week 8, 30 and 54 were 81.0%, 91.3% and 100%, respectively, and clinical remission rates at week 8, 30 and 54 were 38.1%, 47.8% and 50.0%, respectively, while mucosal healing rates at week 8, 30 and 54 were 58.3%, 66.7%, and 66.7%, respectively. We also evaluated the interchangeability of CT-P13 in 27 CD patients and 9 UC patients after switching from its originator. The efficacy of CT-P13 was maintained in 92.6% (25/27) of CD patients and in 66.7% (6/9) of UC patients for at least 32 weeks after switching from its originator. There was no infusion reaction or any serious adverse events related to CT-P13 in these patients.
Another post-marketing study28 included patients with active moderate-to-severe CD, fistulizing CD, or moderate-to-severe UC treated with CT-P13 and followed for 30 weeks. Clinical response rates at week 14 and 30 in patients with moderate-to-severe CD were 87.2% (34/39) and 79.5% (31/39), while clinical remission rates at the same time points were 69.2% (27/39) and 59.0% (23/39), respectively. In the case of fistulizing CD, the clinical response and remission rates were 66.7% (4/6) and 33.3% (2/6) at week 14 and 66.7% (4/6) and 50% (3/6) at week 30, respectively. For patients with moderate-to-severe UC, clinical response and remission rates were 75.5% (40/53) and 49.1% (26/53) at week 14 and 72.2% (39/54) and 37.0% (20/54) at week 30, respectively. Treatment-related adverse events occurred in 10% of patients and were mostly mild-moderate in severity. There were five serious treatment-related adverse events (two infusion-related reactions, two infections, one abdominal pain) and no cases of malignancy, pneumonia, or death.
A recent study29 using 125 IBD patients' and controls' sera demonstrated that anti-Remicade antibodies in IBD patients recognize and functionally inhibit Remsima to a similar degree, suggesting similar immunogenicity profile. All 69 positive anti-Remicade sera were cross-reactive with Remsima. Antibody to infliximab titers against Remicade or Remsima were strongly correlated (r-values between 0.92 and 0.99, P<0.001). Anti-Remicade antibodies of IBD patients exerted similar functional inhibition on Remsima or Remicade TNF-α binding capacity.
CONCLUSIONS
Clinicians should be aware of the benefits and concerns regarding the clinical application of biosimilar in IBD patients. Biosimilars represent an opportunity to reduce healthcare costs even taking into account similarity in terms of efficacy and safety compared with their original products. However, well-designed, prospective randomized non-inferiority trials for efficacy and safety, as well as immunogenicity and interchangeability should be needed to confidently integrate biosimilars into IBD treatment.

 XML Download
XML Download