

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic inflammatory condition in the small and large bowel, and includes two major clinical forms, Crohn's disease (CD) and ulcerative colitis (UC).1 Although the exact causes of IBD are largely unknown, early studies aimed at identifying the genetic factors associated with human IBD revealed nucleotide-binding oligomerization domain-containing protein 2 (NOD2) as a CD-susceptibility gene.2 Ever since NOD2 was introduced, many researchers have tried to find other genetic risk factors for IBD. Based on the efforts of many researchers, a considerable number of genetic risk factors, including solute carrier family 22, member 4 (SLC22A4), SLC22A5, prostaglandin E receptor 4 (PTGER4), interleukin 23 receptor (IL23R), autophagy related 16-like 1 (ATG16L1), immunity-related GTPase family M (IRGM), discs, large homolog 5 (DLG5), and X-box binding protein 1 (XBP1) have been found to be associated with the development of IBD in addition to NOD2.3,4,5 Among them, the genetic association of XBP1 polymorphisms with IBD was suggested by the presence of hypomorphic single-nucleotide polymorphisms in the XBP1 gene in IBD patients, and unabated endoplasmic reticulum (ER) stress was associated with the induction of intestinal inflammation shown in IBD patients.4
Interestingly, several genetic risk factors of IBD were found to be involved in the maturation and function of Paneth cells.6,7 Paneth cells are specialized epithelial cells that reside in the crypts of the small intestine, and produce antimicrobial materials including defensins and lysozymes.8 Reduced antimicrobial function of defensins was reported in IBD patients with a genetic risk factor allele of the NOD2 gene,9 and transcription factor 4 (TCF4), a Wnt transcription factor, is also known to be critical for the proper functioning of Paneth cells and was suggested as another genetic risk factor associated with IBD.10 In addition, XBP1 and ATG16L1 converge in Paneth cells as primary factors of IBD, and ER stress exacerbated by XBP1 deficiency might be attenuated by the compensatory mechanism of autophagy activation in Paneth cells.6
In the inflammatory loci of IBD patients, there is massive infiltration of inflammatory cells including T cells, neutrophils, monocytes, and macrophages.11,12 Recently, the presence of immature myeloid cells, also known as myeloid-derived suppressor cells (MDSCs), was also detected in IBD patients as well as in animal models of IBD.13 In this review, we will discuss the accumulation and functions of MDSCs in IBD-associated intestinal inflammatory conditions, and a possible association of ER stress on the inflammatory or suppressive functions of MDSC's.
MDSCs IN IMMUNE SYSTEMS
MDSCs are a heterogeneous cell population consisting of macrophage precursors, dendritic cells, granulocytes, and early myeloid progenitors.14 The definition of this cell population is based on its origin, myeloid lineage, and its immunosuppressive function. Since the term "MDSC" does not directly reflect the immaturity of this population, they are also called immature myeloid cells.15 They expand during pathological conditions, including infections, inflammations, and cancers in experimental animal models, as well as in human patients.14,16 There are two main subsets of MDSCs: polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (Mo-MDSCs), especially in murine models of tumor transplantation.17,18 In mice, PMN-MDSCs are CD11b+Ly-6G+Ly-6Clow cells, while Mo-MDSCs are CD11b+Ly-6G-Ly-6Chigh cells.19 There are some differences in suppressive capacity and functional mechanism between the two subsets20 and both PMN-MDSCs and Mo-MDSCs can be found in humans.16,21
In the immune system, it is known that the major role of MDSCs is immune suppression, and this seems to be especially true in tumor tissues.15,16 The accumulation of MDSCs in tumor-bearing mice and cancer patients is correlated with poor prognoses. MDSCs can inhibit the proliferation and cytokine production of CD4+ and CD8+ T cells through amino acid deprivation and the release of oxidizing molecules.22 They also induce regulatory T cells to suppress immune responses against tumor cells.23 Besides, they can modulate the function of T cells as well as their migration and viability.22 In addition to T cell inhibition, MDSCs suppressed natural killer (NK) cell cytotoxicity and cytokine production,24 and suppress the antigen presenting functions of dendritic cells.25
Despite their inherent suppressive functions, MDSCs can be converted into immunostimulatory myeloid cells in specific circumstances.26 In acute inflammation, such as trauma and sepsis, MDSCs are the sentinel cells for immune-surveillance and act as immune effectors.27 Even in the ovarian cancer model, MDSCs in ascites play a role as immunostimulatory antigen presenting cells (APCs).28 Furthermore, there is plasticity in the suppressive function of MDSCs. Our recent study also showed that intra-tumoral injection of attenuated Salmonella induced tumor necrosis factor α (TNF-α)-producing PMN-MDSCs, which can function as immune effectors.19 With the help of NKT cells, MDSCs became immunogenic APCs and stimulated antigen specific T cells.29,30 Hence, the divergent roles and the functional plasticity of MDSCs are now interesting subjects in MDSC studies.
MDSCs IN IBD
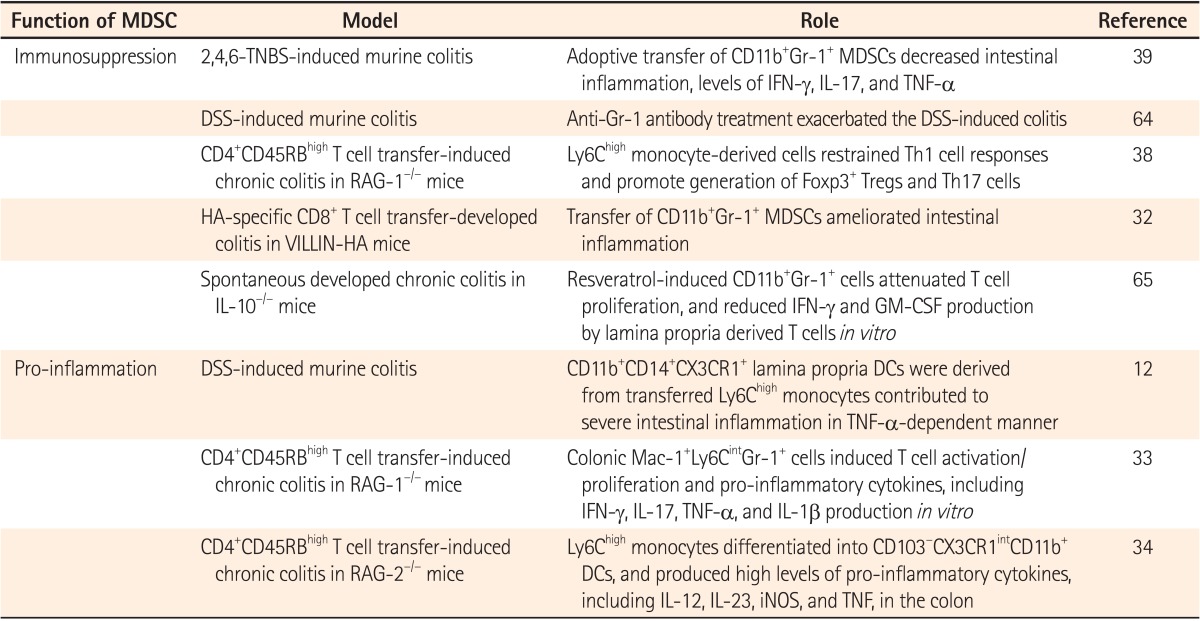
IBD is an autoimmune-associated disease characterized by small and large intestinal inflammation.31 There are two major forms of IBD, CD and UC.1 In the chronic inflammatory condition of IBD, there are complex interactions between several immune cells infiltrating into the intestinal mucosa, with epithelial cells even ignoring the effects of microbiota.11 Among them, myeloid cells, including neutrophils, macrophages, and MDSCs, have been a focus of study due to their divergent role in inflammation.32 In particular, the immunosuppressive function of MDSCs was suggested in several mouse models of IBD (Table 1). It was reported that CD11b+Gr-1+ MDSCs were accumulated in a murine colitis model, and they expressed nitric oxide synthase 2 and arginase, which are known to be critical functional mediators of MDSCs.32 As well as in a model of IBD, CD14+HLA-DRlow MDSCs with suppressive functions were reported to be increased in the peripheral blood of IBD patients.32
Besides MDSCs in IBD models being initially reported as immunosuppressive, as shown in tumor-bearing hosts, the roles of MDSCs during intestinal inflammation have become controversial since recent studies suggested that they functioned as pro-inflammatory myeloid cells. Colonic Mac-1+Ly6CintGr-1+ cells, similar to PMN-MDSCs, acquired stimulatory APC functions and induced T cell activation and pro-inflammatory cytokine production.33 In addition, adoptive transferred Ly6Chi monocytes were converted into pro-inflammatory cells. They then contributed to intestinal inflammation,12 and produced pro-inflammatory cytokines in a murine model of IBD.34
These controversial roles of MDSCs in IBD might be partially due to interleukin 17 (IL-17) cytokine production, which could be at least partially ascribed to increased differentiation of Th17 cells. Th17 cells are known to act as pathogenic effector cells in various immune-related diseases, including experimental autoimmune encephalomyelitis, arthritis, and IBD.13 In the case of IL-17, one study showed that this cytokine had a pathogenic role in severe intestinal inflammation, 35 but another study demonstrated IL-17-mediated protection in murine colitis.36
Interestingly, it was recently reported that CD11b+Gr-1+ MDSCs enhanced Th17 cell differentiation and contributed to the pathogenesis of experimental autoimmune encephalomyelitis. 37 Likewise, a recent study showed that immunosuppressive MDSCs inhibited Th1 responses while they enhanced Th17 generation.38 In a chronic colitis model, pro-inflammatory colonic Mac-1+Ly6CintGr-1+ cells induced IL-17 production by T cells.33 Collectively, this series of studies suggest that MDSCs induced by intestinal inflammation conditions might be involved in Th17 generation and IL-17 production. As described, Th17 cells can contribute to establishing the pro-inflammatory environment and pathogenesis of IBD. On the contrary, MDSCs were initially reported to inhibit the production of several pro-inflammatory cytokines including IL-17, and functioned as immune regulators in several other inflammatory conditions including cancer and chronic infections.39 Thus, the role of MDSCs in IBD is still controversial, and might be closely associated with the production of IL-17 or the induction of IL-17-producing T cells.
ER STRESS AND IBD
ER is a type of organelle in eukaryotic cells responsible for the folding, modification, maturation, and trafficking of newly synthesized proteins, and only properly folded proteins are transported from the rough ER to the Golgi apparatus for secretion.40 Thus, the precise regulation of ER function is critical for the maintenance of cellular homeostasis.41 The condition in which unfolded or misfolded proteins accumulated in the ER is known as ER stress, and can be caused by several factors including disturbances in redox regulation, calcium regulation, glucose deprivation, and viral infection.42 Experimentally, several conditions including ER Ca2+ depletion, defective glycosylation, viral infection, and inflammatory conditions are also known to induce ER stress.43
ER stress activates multiple cellular processes known as unfolded protein responses (UPRs), and three branches of UPRs have been discussed extensively in previous reviews. 42,44 Under ER stress conditions, misfolded or unfolded protein are accumulated in the ER lumen, and trigger UPRs to restore normal ER function. UPR signaling results in the adaptation of cells to ER stress by increasing activation of ER chaperon function, ER trafficking, degradation of ER-resident protein, as well as by inhibiting CAP-dependent translation through phosphorylation of the α-subunit of eukaryotic initiation factor 2 (eIF2α).45 Otherwise, excessive ER stress induces apoptosis via C/EBP-homologous protein (CHOP) and the inositol-requiring transmembrane kinase/endonuclease 1α (IRE1α)/TNF receptor-associated factor 2 (TRAF2)/TNF-α pathway.46 In addition, sustained ER stress also induces cell death due to failure in UPR signaling to induce the homeostatic adaptation of cells.47
In mammalian cells, ER stress evokes UPR to resolve constraint inside the ER.44 UPRs consist of three highly conserved pathways including PKR-like eukaryotic initiation factor 2α kinase (PERK), IRE1, and activating transcription factor-6 (ATF6). Among them, the IRE1a pathway includes splicing of a gene encoding XBP1.4 Spliced XBP1 (sXBP1) is critical for the functions of the ER such as expansion and secretion, especially in several highly secretory cells including plasma cells and pancreatic and salivary gland epithelial cells.48,49 The function of the unspliced form of XBP1 (uXPB1) is not well known, and uXBP1 is highly unstable.4 Instead, sXPB1 triggers the transcription of genes for the quality control and maintenance of ER functions. IRE1 is an ancestrally conserved pathway of UPR, and also possesses kinase activity to phosphorylate TRAF2 as well as endonuclease activity. 4 Accordingly, intestinal epithelial cell-specific depletion of XBP1 is known to initiate spontaneous small intestinal inflammation in mice.
UPR is known to intersect with several inflammatory pathways including IκB kinase (IKK) and c-Jun N-terminal kinases (JNK),50,51 and is also associated with inflammation in obese and diabetic patients.52 In addition, ER stress has been associated with the development of IBD.4 In humans, several genes associated with ER stress including XBP1, anterior gradient 2 (AGR2), mucin 19 (MUC19), and ORMDL sphingolipid biosynthesis regulator 3 (ORMDL3) have been reported as genetic risk factors for IBD, and increased ER stress has been observed in patients with IBD.5 In several mouse models, ER stress induced by deletion of some ER chaperones such as ATF6 or p58IPK led to the development of severe intestinal inflammation after dextran sulfate sodium (DSS) administration,53 and the administration of chemical chaperones including 4-phenylbutyrate and tauroursodeoxycholate ameliorated colitis in those mice.53 In addition, conditional knockout of XBP1 in intestinal epithelial cells causes small intestinal enteritis with crypt abscesses reminiscent of human IBD,4 and AGR2 knockout mice develop granulomatous ileocolitis.54 Interestingly, UPR signaling was found to be impaired in colonic epithelial cells of protein kinase RNA-activated (PKR)-/- mice with a reduced level of phosphorylated eIF2α and diminished ER chaperons, resulting in more severe DSS-induced colitis in these mice.55 In addition, glutamine treatment attenuates ER stress increased in a trinitrobenzene sulfonate (TNBS)-treated colitis model through modulation of UPR signaling, and diminishes the severity of macroscopic damage and apoptotic cell death.56
ER STRESS AND THE DIVERGENT FUNCTIONS OF MDSCs
In tumor-bearing hosts, it has been reported that ER stress is transmitted from tumor cells to myeloid cells.57 In association with increased inflammation by ER stress, ER stress-conditioned medium obtained from tumor cells culture with ER stress inducers resulted in up-regulation of ER stress in myeloid cells. Similarly, infectious ER stress has also been reported in tumor cells, which was known to be mediated by Par-4 secreted by ER stressed cells.58 Hypoxic tumor micro-environments could induce an ER stress condition, as well as increased reactive oxygen species generated in MDSCs induced ER stress. Increased ER stress results in early apoptosis of MDSCs in a TNF-related apoptosis-inducing ligand-receptor (TRAIL-R)-dependent manner, which in turn accelerates the generation of MDSCs in bone marrow. Indeed, MDSCs found in tumor-bearing mice and cancer patients showed significantly higher levels of ER stress compared with healthy controls.59 However, it is not clear whether ER stress increased in IBD condition influences on intestinal MDSCs found in IBD patients as well as on their immuno-suppressive or inflammatory functions.
Although aggravation of inflammation by ER stress seems to be certain in several disease conditions, the mechanism by which ER stress triggers inflammation remains largely unknown. IRE1/TRAF2-mediated JNK activation and the nuclear factor-κB (NF-κB) pathway could explain the inflammatory responses observed in the ER stress condition. ER stress also activates cleavage of ATF6 to activate the expression of acute-phase protein genes in the liver, and mediates acute inflammatory responses.60 Recent studies have also suggested the role of the nucleotide-binding oligomerization domain, leucine rich repeat, and pyrin domain containing 3 (NALP3) inflammasome in chronic inflammatory disease with ER stress.61 Unregulated activation of caspase-1 and subsequent overproduction of IL-1β through inflammasome activation might be associated with the perpetuation of IBD. Indeed, there is an increased level of IL-1β in the intestinal specimens of IBD patients.62 On the contrary, loss of function in NOD2 or inflammasomes could result in dysbiosis by decreased IL-1β.63 Thus, the involvement of several genetic risk factors, including NOD2, ATG16L1, NALP3, and chemokine (C-C motif) receptor 6 in the inflammatory status of MDSCs need to be further elucidated.
CONCLUSIONS
ER stress responses in MDSCs as well as in intestinal epithelial cells might be critical for the homeostatic regulation of gut immunity. In this review, we focused on the roles of MDSCs on intestinal inflammation, especially in animal models of IBD and also in IBD patients. The recent advances in experimental techniques for intestinal tissues, wide availability of germ-free mice, genome-wide analysis of patients' genetic information, and metagenomic analysis of commensals increase the understanding of the development and progress of IBD, and hence provide some clues for the development of therapeutic drugs for the treatment of IBD. In particular, some strategies to regulate ER stress responses in MDSCs as well as in the intestinal epithelium might be novel ways to prevent or treat intestinal inflammation in IBD patients.
 XML Download
XML Download