

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a member of the tumor necrosis factor family of cytokines. The binding of TRAIL to its receptors, death receptor 4 (DR4) and DR5, triggers apoptotic signaling. The activation of DR4 or DR5 recruits the Fas-associated death domain protein (FADD) and procaspase-8 to form the death-inducing signaling complex (DISC), which leads to the activation of the caspase cascade.1,2 Caspase activation can be suppressed by the inhibitor of apoptosis protein (IAP) family members, as well as by anti-apoptotic B-cell lymphoma 2 (Bcl-2) family proteins.3 TRAIL has been reported to trigger selective and specific apoptotic cell death events in vitro and in vivo, with no significant side effect on normal cells.2,4 The use of TRAIL in such therapy, is currently undergoing intensive clinical evaluation. Although TRAIL is a potent anti-tumor agent in preclinical models, it has also been reported that some tumor cells intrinsically process or acquire resistance to TRAIL. TRAIL resistance can occur at several steps in the signaling cascade and can arise through different mechanisms. In colorectal cancer (CRC) cells, several mechanisms contributing to TRAIL resistance have been reported, including deficient receptor redistribution to the membrane,5 mutation of caspase-8,6 cellular fas-associated death domain-like interleukin-1-β-converting enzyme-inhibitory protein (cFLIP) expression,7 Bax deficiency,8 or through X-linked inhibitor of apoptosis protein (XIAP) expression.9 As a result, it is not recommended to use TRAIL as a single agent. Combined treatment with chemotherapeutic drugs has been shown to overcome TRAIL resistance in many cancer cell types.10-12
Parthenolide (PT), a naturally occurring agent, has been used for the treatment of fever and inflammatory disease.13 Over the past two decades, it has been established that PT induces apoptosis in various cancer cell types, including human hepatocellular carcinoma cells, human lung and stomach cancer cells, and glioblastoma cells.10,14-17 It has been shown that the apoptotic effect of PT is associated with the inhibition of nuclear factor κB (NF-κB),18 and signal transducer and activator of transcription 3 (STAT3),18 enhanced oxidative stress,19 and activation of the mitochondria-mediated pathway.20 In our previous study, we found that PT effectively induced apoptosis in CRC cells by causing mitochondrial dysfunction in vitro and in vivo.21
In recent years, the use of PT in combination therapy has been investigated in several studies. PT reportedly sensitizes cancer cells to NSAIDs, anticancer drugs, and radiation.22-26 In addition, we previously showed that a combination of PT and 5-fluorouracil (5-FU) can overcome 5-FU resistance in human CRC cells.27 Although it has been reported that PT sensitizes TRAIL resistant hepatocellular carcinoma and breast cancer cells,10,28 it has not been examined whether combined treatment with PT can sensitizes TRAIL resistance in CRC cells.
In this study, we investigated whether PT sensitized CRC cells to TRAIL-induced apoptosis. We hypothesized that PT, in combination with TRAIL, would inhibit proliferation and induce apoptosis in CRC cells. Thus, our objective was to evaluate combination therapy with PT and TRAIL as a potential treatment for TRAIL-resistant CRC.
METHODS
1. Chemicals and Reagents
PT and z-VAD-FMK were purchased from Calbiochem (San Diego, CA, USA). TRAIL was purchased from Peprotech (Rocky Hill, NJ, USA). PT was dissolved in dimethylsulfoxide (DMSO; Sigma, St. Louis, MO, USA) to a concentration of 100 µM and stored in the dark at -20℃. Annexin-V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) were purchased from Invitrogen (Eugene, OR, USA). Hoechst 33258 was purchased from Sigma (St. Louis, MO, USA). Anti-Bcl2, anti-Bid, anti-Bax, anti-cytochrome C, anti-p53 and anti-caspase 3 antibodies were purchased from Santa Cruz Technology (Beverly, MA, USA). Anti-cFLIP and anti-caspase-9 antibodies were purchased from Cell Signaling (Beverly, MA, USA). Anti-actin antibody was purchased from Sigma.
2. Cell Culture and Treatment
The human CRC cell lines, HT-29 and HCT 116 cells (American Type Culture Collection, Rockville, MD, USA), were used as TRAIL-resistant and TRAIL-sensitive cells, respectively. The cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 units of penicillin, and 100 units of streptomycin. For PT or TRAIL treatment, cells were sub-cultured in RPMI 1640 medium without FBS for 12 hours. PT and TRAIL were diluted with FBS-free medium to achieve desired concentrations. DMSO was diluted to the equivalent concentration and applied to the cells as a control.
3. Cell Viability Assay
Human CRC cells were plated at a density of 1.0×104 cells per well in 96 well plates. Cells were treated with PT and/or TRAIL for 24 hours, before the medium was removed from each well and replaced with 200 µL of fresh medium plus 20 µL of 3-(4, 5-dimethylthiazol-2yl)-2, 5-diphenyltetrazolium bromide (MTT, 2.5 mg dissolved in 50 µL of DMSO). After incubation for 4 hours at 37℃, the culture medium containing MTT was removed and 200 µL of DMSO was added. Plates were placed on a shaker until the crystals dissolved. Viable cells were detected by measuring absorbance at 570 nm, by using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
4. Annexin-V-fluorescein Staining
After 24 hours of treatment, cells were trypsinized, collected, washed with ice-cold phosphate-buffered saline (PBS), suspended in a 500 µL annexin V binding buffer containing 5 µL of annexin V-FITC, and incubated for 15 minutes at room temperature in the dark. The fluorescence was measured using a BD LSR flow cytometer (BD Biosciences, San Jose, CA, USA) and processed with Cell Quest software for analysis.
5. Cell Cycle and Sub-G1 Analysis
Cell cycle phase and sub-G1 distribution were determined by staining DNA with PI (Sigma-Aldrich), a fluorescent biomolecule (Ex/Em=488 nm/617 nm). In brief, 1×106 cells were incubated with one or both agents for 24 hours. Cells were then washed with PBS and fixed in 70% ethanol overnight. Cells were washed again with PBS and then incubated with PI (10 µg/mL) and simultaneously treatment with RNase at 37℃ for 1 hour. The percentage of cells in different phases of the cell cycle or having sub-G1 DNA content was measured with a BD LSR flow cytometer and analyzed using Cell Quest software.
6. Hoechst 33258 Staining
Apoptosis was identified by the presence of DNA condensation in Hoechst 33258-staining cells. The cells were treated for 24 hours, and then stained with Hoechst 33258 (1 µg/mL) at 37℃ for 10 minutes. To identify cells undergoing apoptosis, nuclear morphology was examined using confocal laser scanning microscope (Carl Zeiss, Jena, Germany).
7. Cell Extraction and Western Blotting
After 24 hours of treatment, cells were collected, washed twice with PBS, and then lysed for 30 minutes on ice, in lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM EDTA, 1% TritonX-100, 0.5% sodium dodecyl sulfate [SDS] and protease inhibitor cocktail). The protein concentrations in the cell lysates were measured using the Protein Quantification kit from Bio-Rad. 50 µg of total protein was loaded onto a SDS-PAGE gel. After transferring and blocking, the membrane was probed with the following antibodies (anti-Bcl2, anti-Bid, anti-Bax, anti-cytochrome C, anti-p53, anti-caspase-3, anti-caspase-9, anti-cFLIP and anti-actin). The signal was detected using enhanced Westone (Intron, Daejeon, Korea) and analyzed using the Las-3000 luminescent Image Analyzer (Fuji Film, Tokyo, Japan).
8. Statistical Analysis
The data are presented as mean±standard error (SE) of at least three independent experiments done in duplicate. Representative Western blots are shown. All the data was entered into Microsoft Excel 5.0, and SPSS Software was used to perform two-tailed t-tests or analysis of variance. P values<0.05 were considered significant.
RESULTS
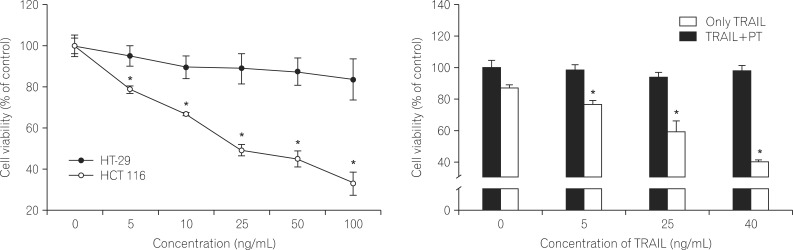
1. PT Enhances the Effect of TRAIL on the Viability of Human CRC Cells
The human colorectal cancer cell lines HT-29 and HCT-116 were treated with TRAIL at various concentrations (0, 5, 10, 25, 50 or 100 ng/mL). After 24 hours of treatment, cell viability was detected using the MTT assay. Treatment of HT-29 cells with TRAIL alone (100 ng/mL) decreased cell viability by approximately 15% (Fig. 1A). In contrast, treatment of HCT 116 cells with TRAIL (100 ng/mL) dramatically reduced viability in a dose-dependent manner, with cell showing a 70% decrease in viability. This indicates that HT-29 cells are highly resistant to TRAIL-induced cell death.
To determine the synergistic effect of PT on TRAIL-induced cell death, HT-29 cells were incubated in the absence or presence of PT (10 µM) and TRAIL (5, 25, or 40 ng/mL). TRAIL alone did not inhibit cell survival (less than 10%), whereas combined treatment with PT exhibited a does dependent decrease in cell viability (Fig. 1B).
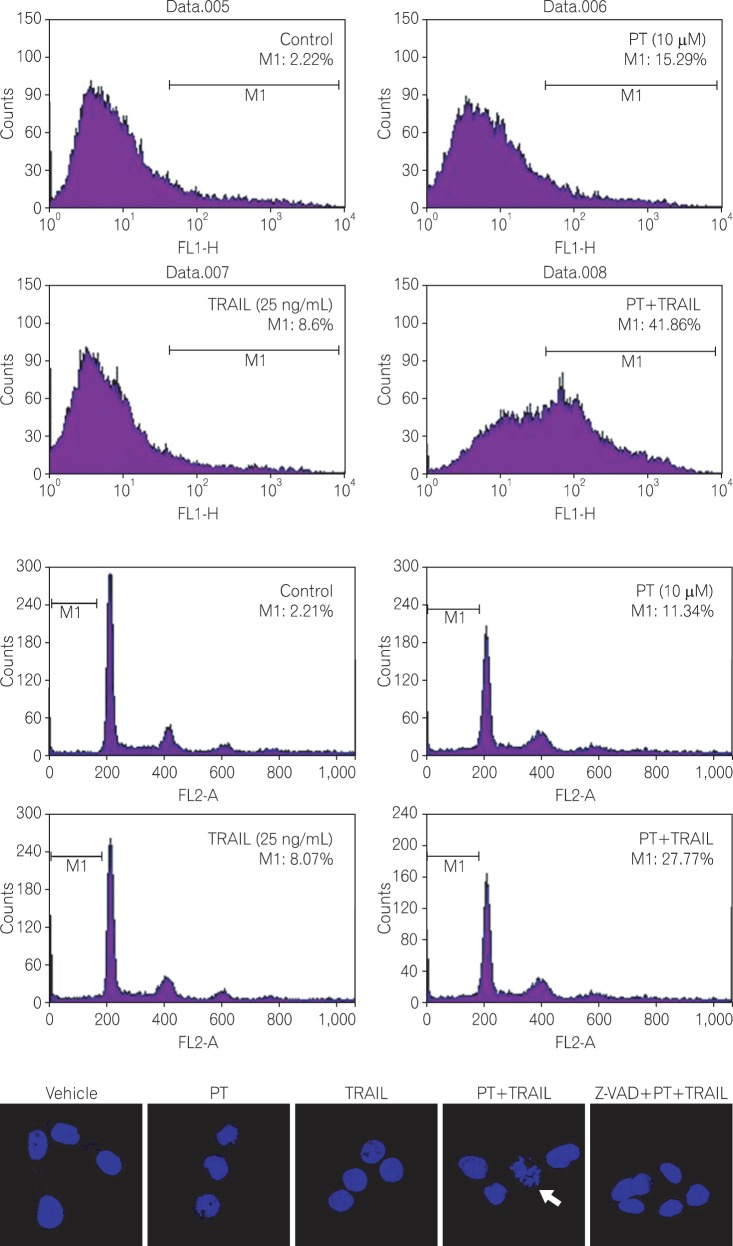
2. PT Enhances TRAIL - induced Apoptotic Cell Death
To support the earlier observations, annexin-V analysis was performed using FACScan. As shown in Fig. 2A, approximately 15.29% of HT-29 cells treated with PT were annexin V-positive, a value comparable to that of TRAIL-treated cells (8.6%). Co-treatment with TRAIL and PT caused a 3-fold increase in the proportion of annexin V-positive cells (41.86%), indicating that PT promotes TRAIL-induced apoptosis in TRAIL-resistant cells.
We also evaluated cell cycle modifications induced by PT and TRAIL in HT-29 cells. 24 hours after incubation with one or both agents, cells were analyzed by PI staining and flow cytometric analysis. Treating cells with PT and/or TRAIL resulted in the presence of a sub-G1 population, indicating apoptotic cell death. Peaks accounting for 11.34% and 8.07% of the overall cell population were detected in cells treated with PT or TRAIL, respectively. In combined treatment, a much greater sub-G1 population (27.77%) was observed, indicating that the combination of two agents dramatically promoted apoptosis in TRAIL-resistant cells (Fig. 2B).
Next, cells were stained with Hoechst 33258 and visualized by confocal microscopy to determine the presence apoptotic nuclear morphology. After treatment with either PT or TRAIL alone, cells were regular in morphology and formed confluent colonies, with cells rarely sloughing off. In contrast, upon treatment with both agents, HT-29 cells exhibited apoptotic characteristics, including cell shrinkage, nuclear condensation, and nuclear fragmentation. Moreover, the pan-caspase inhibitor Z-VAD-FMK blocked the nuclear fragmentation and condensation induced by the combination treatment, indicating that the change in nuclear morphology is mediated by the activation of caspase (Fig. 2C).
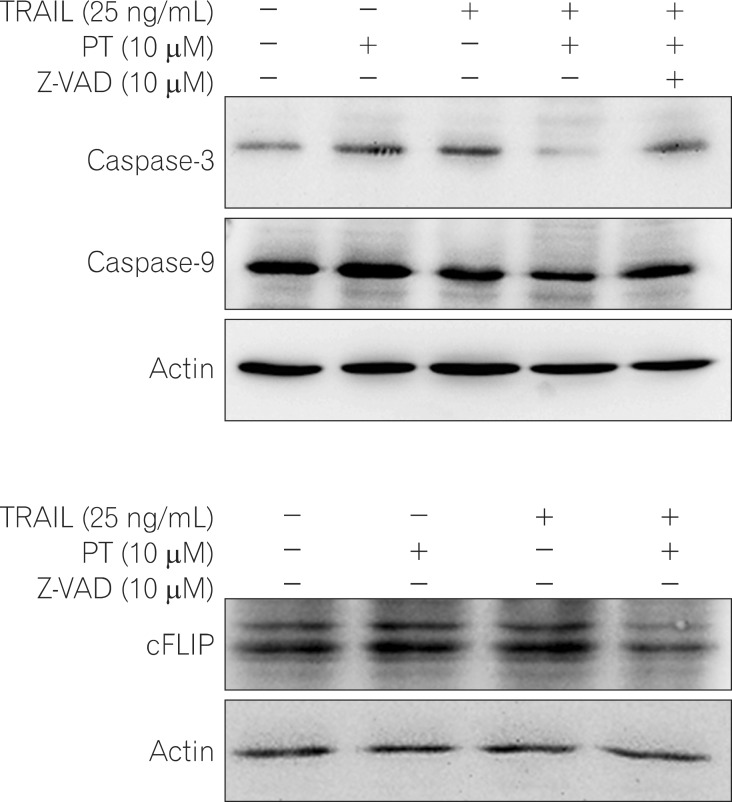
3. PT enhances TRAIL-induced Apoptotic Via Caspase Activation
Many anticancer agents are capable of initiating caspase activation and inducing apoptotic cell death.29 The effect of PT, TRAIL, or PT plus TRAIL treatment on caspase activation in TRAIL resistant cells was examined. Western blot analysis revealed that the levels of full length caspase-3 and -9 in cells co-treated with PT plus TRAIL were significantly decreased compared to those in cells treated with PT or TRAIL alone. Furthermore, pretreatment with pan-casepase inhibitor Z-VAD-FMK significantly blocked the decrease in caspase-3 and -9 levels induced by co-treating cells with PT and TRAIL. These data indicate that co-treatment with PT and TRAIL activates caspases, promoting their enzymatic activity (Fig. 3A).
cFLIP inhibits the activation of caspase-8, the factor that initiates apoptosis.30 Thus, we investigated whether treatment with PT and TRAIL affected the levels of cFLIP proteins in HT-29 cells. Relative to the level of cFLIP detected in cells treated with either PT or TRAIL alone, the level of cFLIP decreased in response to combined treatment (Fig. 3B).
4. PT Enhances TRAIL- induced Expression of Apoptosis Associated Proteins
To evaluate the mechanisms responsible for apoptosis induced by the combination of TRAIL and PT, we investigated the expression level of several pro-apoptosis and anti-apoptosis proteins. In comparison to cells treated with PT or TRAIL alone, Western blot analysis showed that the level of Bcl-2 was significantly decreased by co-treatment. In contrast, the level of expression of truncated-Bid and Bax were significantly increased by combined treatment with PT and TRAIL when compared to those found in cells treated with PT or TRAIL alone (Fig. 4; first, second and third panel).
A consequence associated with decreased Bcl-2 levels is the dissipation of mitochondrial potential and the release of the mitochondrial pro-apoptotic protein, cytochrome C into the cytosol. After combined treatment with PT and TARIL, the release of cytochrome C was increased relative to control-treated cells and cells treated with PT or TRAIL alone (Fig. 4; fourth panel).
In addition, the p53 gene, a hallmark of apoptosis, is inactivated in the majority of human cancers.31 When compared to PT or TRAIL treatment alone, combination treatment significantly increased the level of p53 (Fig. 4; fifth panel). Taken together, these results indicate that the apoptosis induced by the combination of TRAIL and PT may be associated with the mitochondrial pathway.
DISCUSSION
Combination drug therapies have played a prominent role in cancer therapy. Exploration of the molecular mechanisms underlying the synergistic effects achieved by various chemotherapeutic agents would useful developing combination therapeutics that could significantly improve the prognosis of cancer patients.
In this study, we investigated the consequences of TRAIL and PT combination treatment. This treatment resulted in increased apoptosis and caspase-activation, and enhanced TRAIL sensitivity in TRAIL-resistant CRC cells. Therefore, the combination of TRAIL and PT might overcome TRAIL resistance in CRC cells and be an effective therapeutic strategy for patients with CRC.
Two previous studies have demonstrated that PT sensitizes cells to TRAIL. In breast cancer cells, it has been shown that PT attenuates resistance to TRAIL-induced apoptosis.28 PT was also found to sensitize hepatocellular carcinoma cells to TRAIL-induced apoptosis.10 Based on these studies, we hypothesized that PT is a promising candidate for combination therapy with TRAIL in CRC. To support our hypothesis, we first examined the sensitivity of human CRC cell lines to TRAIL. As shown in Fig. 1A, TRAIL inhibited proliferation of HCT 116 cells in a dose-dependent manner; notably, however, this reduction did not occur in TRAIL-resistant HT-29 cells, even at higher doses of TRAIL. These results confirmed that HT-29 cells are resistant to TRAIL and are consistent with the results of previous studies exploring the sensitivity of human CRC cells to TRAIL treatment.32,33 Interestingly, the combined treatment with PT and TRAIL reduced cell proliferation and increased apoptotic cell death in TRAIL-resistant HT-29 cells.
Many factors for apoptosis converge to activate caspase-3-the key protease and final effector in the apoptosis pathway.34 The caspase-dependent apoptosis pathway includes the mitochondrial, death receptor, and endoplasmic reticulum (ER) pathways.35,36 The mitochondrial pathway is regulated by the Bcl-2 family, which is divided into two groups: the anti-apoptotic members (Bcl-2, Bcl-xl)36,37 and pro-apoptotic members (Bax, Bad, Bid).38 In this study, combined PT and TRAIL treatment altered the levels of Bcl-2, truncated-Bid, Bax, caspase-9, and caspase-3. Moreover, the decrease of both caspase-9 and -3 was prevented by pretreatment with a pan-caspase inhibitor, Z-VAD-FMK. These results demonstrate that combined PT and TRAIL treatment circumvent TRAIL resistance in CRC cells. Furthermore, these data suggest that apoptosis induced by combined PT and TRAIL treatment is under the control of a caspase- and mitochondrial pathway.
The canonical tumor suppressor gene, p53, mediates G1 growth arrest by inducing the cyclin-dependent kinase inhibitor p21, and participates in apoptosis through transactivation of the pro-apoptotic Bax gene in response to DNA-damage.39,40 The p53 gene, which is inactivated in a majority of human cancers,31 has been proposed as an accurate indicator that CRC is responding to anticancer drugs. In this study, we demonstrated that the level of p53 was enhanced by combined PT and TRAIL treatment.
A structural homologue of caspase-8, the anti-apoptotic factor cFLIP binds to FADD, competing with caspase-8 for recruitment to DISC. The overexpression of cFLIP in tumor cells has been shown to determine the resistance of a tumor to TRAIL.41,42 Importantly, aberrant up-regulation of cFLIP proteins is present in a number of cancers. In addition, down-regulation of cFLIP by small interfering RNA is sufficient to sensitize TRAIL-resistant tumor cell lines to TRAIL-induced apoptosis.43,44 Although we did not show the change in caspase-8 expression, the downregulation of cFLIP was observed in HT-29 cells under combined treatment conditions, implicating the involvement of reduced cFLIP levels in this process.
In summary, we investigated whether combined PT and TRAIL treatment could inhibit cell proliferation and induce apoptotic cell death in TRAIL-resistant CRC cells. Combined treatment with PT and TRAIL upregulates pro-apoptotic proteins and downregulates anti-apoptotic proteins, leading to the release of cytochrome C, increased caspase activation, and subsequently, increased apoptosis. Taken together, these results suggest that PT sensitizes cells to TRAIL via mitochondrial- and caspase-dependent apoptotic pathways in human colorectal cancer cells. For this reason we believe that combined treatment with PT and TRAIL could represent a new and important therapeutic strategy for CRC treatment.

 XML Download
XML Download