

 PDF
PDF Citation
Citation Print
Print



Go to : 
INTRODUCTION
Atypical hemolytic uremic syndrome (aHUS) is a form of thrombotic microangiopathy (TMA) characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury [1]. aHUS stems from a genetically inherited or acquired dysfunction of complement proteins, which results in the upregulation of the alternative complement pathway. aHUS is usually diagnosed as a sporadic form that is triggered by events such as pregnancy, autoimmune conditions, organ transplantation, malignancy, and the use of certain drugs [1].
Posttransplant TMA usually develops de novo and is provoked by factors such as ischemic-reperfusion injury, antibody-mediated rejection (AMR), viral infection, or use of immunosuppressants [2,3]; however, a significant proportion of patients with de novo, posttransplant TMA have predisposing genetic factors associated with complement dysregulation and are diagnosed with aHUS [4]. These findings suggest blockade of the complement cascade can be used to treat posttransplant TMA refractory to conventional plasma exchange (PE) therapy. Several case studies of posttransplant TMA have reported treatment responses to eculizumab, a humanized monoclonal antibody directed against the terminal complement protein C5 [5-7]. In this case report, we describe the first successful eculizumab rescue therapy case of a kidney transplant recipient with aHUS in South Korea.
Go to :
CASE REPORT
This study was approved by the Institutional Review Board of Severance Hospital (IRB No. 4-2022-1091). Informed consent from the patient was taken.
A 32-year-old man with end-stage renal disease caused by immunoglobulin A nephropathy underwent preemptive robotic living donor kidney transplantation (KT). The donor was the patient’s 60-year-old mother. Preoperative complement-dependent cytotoxicity and a flow cytometric cross-match test were negative; there were no donor-specific anti-human leukocyte antigen antibodies (DSA). The transplanted kidney showed good reperfusion and functioned immediately with sufficient intraoperative urine output. Antithymocyte globulin (ATG) was used as an induction agent, along with tacrolimus, mycophenolate mofetil (MMF), and methylprednisolone.
Urine output was unremarkable between postoperative days (PODs) 0 and 2, yet the patient’s blood pressure levels were elevated, ranging from 148/97 mmHg to 184/114 mmHg; the platelet count dropped from 247×103/μL to 57×103/μL. On POD 3, a clinical diagnosis of TMA was made based on the platelet count (as low as 34×103/μL), low hemoglobin (Hb) levels of 7.9 g/dL without evidence of bleeding, highly elevated lactate dehydrogenase (LDH) levels (1,145 IU/L), decreased haptoglobin levels (<5 mg/dL), and positive schistocytes on a peripheral blood smear.
Serum creatinine (sCr) levels remained high (5.02 mg/dL) compared to a preoperative level of 7.88 mg/dL. C3 levels decreased to 67.7 mg/dL, while C4 levels were within the normal range. The tacrolimus trough level was 4.7 ng/mL, and serum DSA levels were negative. PE was immediately initiated, and laboratory analysis of a disintegrin and metalloproteinase with thrombospondin type-1 motif 13 (ADAMTS13), identification of enterohemorrhagic Escherichia coli (EHEC) toxin by polymerase chain reaction, and next-generation sequencing (NGS) of genes related to aHUS and coagulopathy were promptly performed. Tacrolimus was discontinued because of concerns about toxicity. ATG was administered on PODs 1 and 4–7 (1.5 mg/kg/day on PODs 1, 4, and 5; 1 mg/kg/day on PODs 6 and 7). Steroid pulse therapy was performed on PODs 3 and 4 (500 mg and 250 mg of methylprednisolone, respectively) to prevent acute rejection. Changes in Hb, platelet, sCr, and LDH levels during PE and the hospitalization period are shown in Figure 1A and B. Two cycles of hemodialysis were needed during PE treatment. A renal biopsy performed after the fifth PE treatment suggested TMA, with the presence of CD61 in the glomerular capillary lumen, peritubular capillaries, and arteriolar wall (Fig. 2A and B). There was no evidence of acute rejection, and C4d staining was negative. Laboratory tests revealed an ADAMTS13 activity of 24% and were negative for EHEC toxin; polyspecific, anti-C3, or anti-immunoglobulin G antibodies; or infectious causes, including cytomegalovirus or BK viremia.
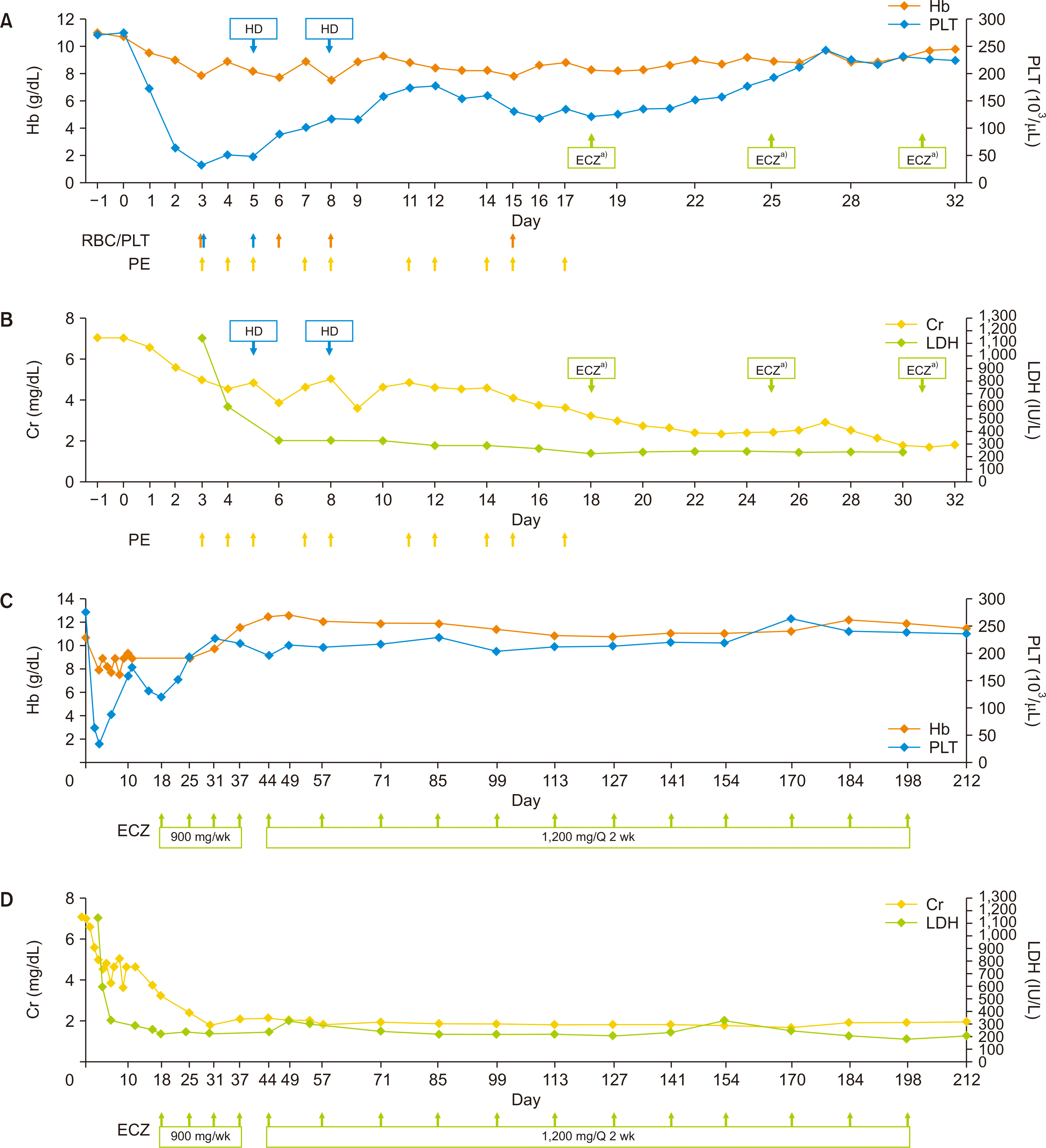 | Fig. 1Clinical course of the indicated laboratory values after kidney transplantation, during in-hospital days (A, B), and during eculizumab administration (C, D). Transfusion of red blood cells (RBCs; orange arrows) or platelets (PLT; blue arrows), each plasma exchange (PE) session (yellow arrows) and eculizumab (ECZ) treatment (green arrows) are depicted under each figure and denoted with an arrow. Hb, hemoglobin; HD, hemodialysis; Cr, creatinine; LDH, lactate dehydrogenase; Q, quisque. a)ECZ, 900 mg.
|
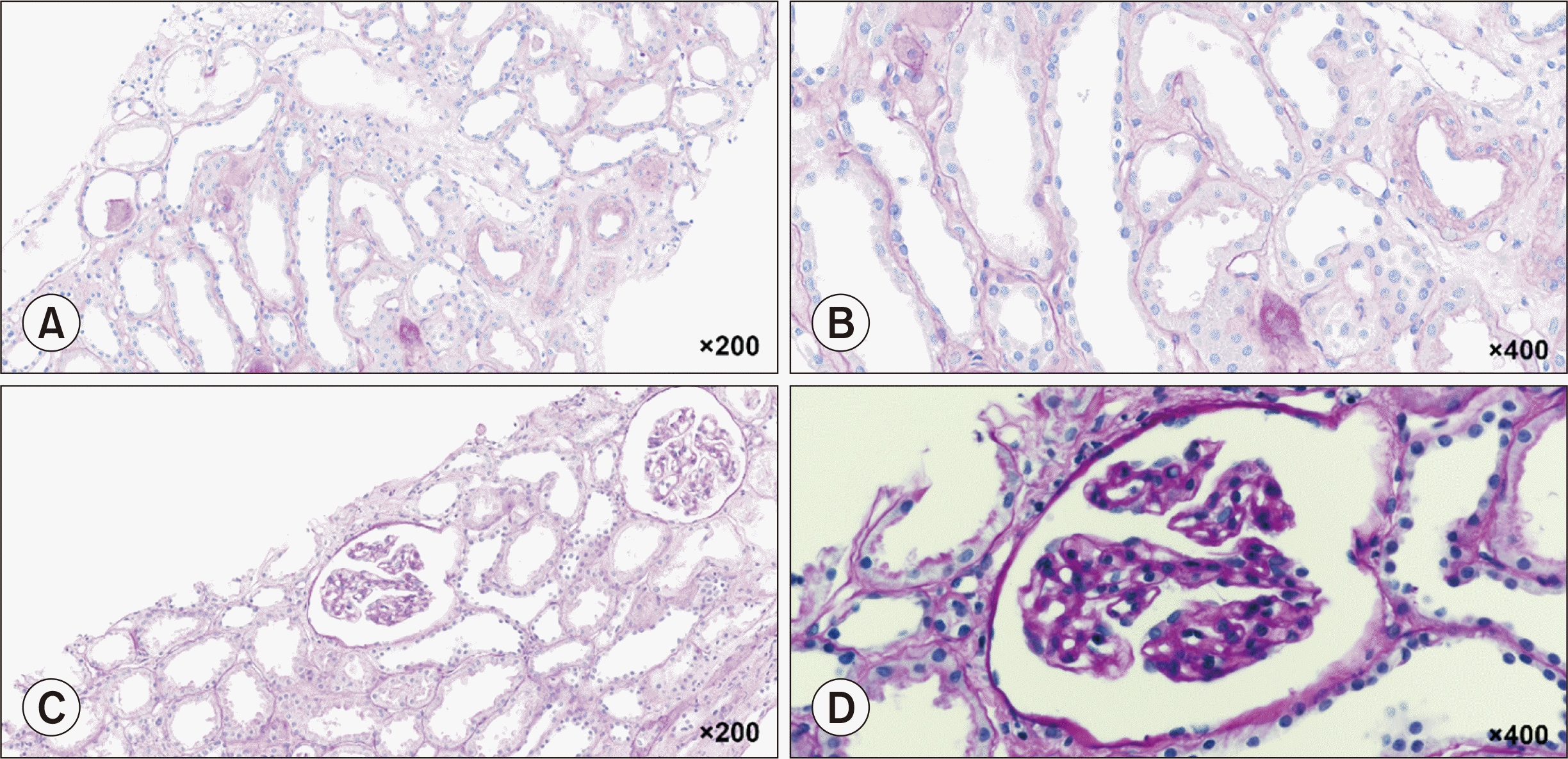 | Fig. 2Periodic acid-Schiff (PAS) staining of posttreatment allograft biopsies. (A, B) A stained section on postoperative day (POD) 10 following the fifth plasma exchange (PE) treatment. The tubules show diffuse acute tubular injury with rare mitotic figures. Minimal tubular atrophy is present. The blood vessels show focal-prominent endothelial cells, consistent with thrombotic microangiopathy (TMA) (PAS; ×200 [A], ×400 [B]). (C, D) Stained section on POD 28 after the tenth PE treatment and two doses of eculizumab. The glomerular basement membrane is mildly thickened with a double contour, suggestive of chronic TMA (PAS; ×200 [C], ×400 [D]).
|
Due to a partial hematological response and persistent renal dysfunction despite 10 PE sessions (sCr level of 3.25 mg/dL), eculizumab therapy (900 mg weekly) was initiated on POD 18 because of suspected aHUS. Meningococcal vaccination was performed on POD 5, in anticipation of eculizumab treatment.
On POD 22, NGS results revealed gene mutations classified as variants of unknown significance (VOUS) in coagulation-associated genes, including PROC, NBEAL2, A2M, C4A, TBXAS1, and GATA1. A second renal biopsy performed on POD 28 after the tenth PE treatment and two doses of eculizumab revealed mild focal thickening of the glomerular basement membrane, suggesting chronic TMA, without evidence of rejection (Fig. 2C and D). Laboratory data gradually improved after three doses of eculizumab, and the patient was discharged on POD 33 with an sCr level of 1.82 mg/dL (Fig. 1C and D). In total, 16 doses of eculizumab were administered for 27 weeks (900 mg weekly for 4 weeks, 1,200 mg during week 5, and 1,200 mg every 2 weeks thereafter).
At 8 months after the last dose of eculizumab, the patient’s sCr levels abruptly increased to 3.28 mg/dL from a median sCr level of 2.00 mg/dL. Maintenance immunosuppression consisted of a sirolimus level of 8.7 ng/mL, MMF, and prednisolone. The laboratory findings were inconsistent with TMA. A renal biopsy showed acute tubular injury with CD61 positivity, presumably from the previous TMA. Because of suspected chronic TMA, two cycles of PE and intravenous immunoglobulin (0.2 g/kg) were administered. sCr levels improved to 2.14 mg/dL. At the last follow-up 27 months after KT, the kidney was functioning well with an sCr level of 1.83 mg/dL. Hb, platelet, and LDH levels were all within the normal range.
Go to :
DISCUSSION
De novo TMA after transplantation occurs in 0.8%–14% of patients with renal allografts; an approximate graft loss rate of 30%–40% within 2–3 years posttransplant has been reported [8]. Here, we reported a case of de novo TMA after KT caused by aHUS. Normal ADAMTS13 activity excluded a diagnosis of thrombotic thrombocytopenic purpura. There was no evidence of active AMR or infectious causes. Dysregulation of the alternative complement pathway was suspected based on low C3 levels and persistent clinical features of TMA after PE. The rapid response to eculizumab and resolution of TMA demonstrated an underlying complement pathway dysfunction; thus, a diagnosis of aHUS was appropriate.
In about 50% of patients with aHUS, variations in complement protein genes, such as CFH, MCP, CFI, C3, and C3B, and non-complement pathway members, such as THBD and DGKE, drive complement dysregulation [9,10]. Nevertheless, significant levels of gene variants are not detected in most patients with a clinical diagnosis of aHUS who respond to eculizumab [6,11], including the patient in our case. In our study, NGS did not reveal any of the above-mentioned gene variants, while mutations in coagulation-associated VOUS were detected, including PROC (the gene encoding protein C). Protein C is a plasma vitamin K-dependent protease with anticoagulant properties that functions by inactivating factor (F) VIIIa and FVa, cofactors in the activation of FX and prothrombin, respectively. Mutations of a single copy of PROC in a heterozygous individual have been shown to cause various degrees of low protein C activity and thrombophilia, triggered by clinical risk factors [12]. According to the work of Amara et al. [13], the coagulation and complement systems each control their own activation status, in which FXa and thrombin have been found to cleave C3 and C5. Therefore, decreased levels of protein C activity and a triggering condition that enhances the activation of both FXa and thrombin may contribute to the dysregulation of the alternative complement pathway. This scenario provides one mechanism by which the therapeutic effects of eculizumab manifested in our case.
Regarding the time to begin eculizumab treatment in patients with de novo TMA after KT, earlier use seems favored. A study involving 22 patients with de novo post-KT aHUS reported a shorter median time (5 days) to start eculizumab resulted in a complete response compared to later initiation of treatment (22 days), which resulted in either partial or no response [5]. In our case, eculizumab treatment was promptly started 15 days after the TMA diagnosis, when platelet levels decreased after the initial response to PE sessions. For discontinuation of eculizumab in patients with aHUS, a case-by-case approach after at least 6 to 12 months of treatment and at least 3 months of kidney function normalization/stabilization is recommended [14]. However, there are no definitive criteria outlining the duration of eculizumab treatment in aHUS developing after KT [15]. Considering the possibility of coagulation system dysregulation resulting in sustained activation of complement in our patient, we decided to withdraw the drug when a stabilized sCr level and hematologic normalization were maintained for 3 months.
This is the first case of successful eculizumab rescue therapy for post-KT aHUS in South Korea. At the time of diagnosis, the use of eculizumab in aHUS after organ transplantation was not reimbursed by the National Health Insurance System of Korea; therefore, a detailed review was required for approval. In this case, the rapid diagnosis of aHUS enabled reimbursement approval, and early treatment led to the rescue of allograft dysfunction. In addition, the patient discontinued eculizumab successfully, maintaining graft function during long-term follow-up.
To summarize, we demonstrated a case of aHUS diagnosed after KT that was rescued by the early administration of eculizumab upon partial response to conventional therapy. De novo TMA after KT can occur because of the activation of the complement pathway. Early eculizumab treatment appears important for the successful treatment of aHUS that is refractory to PE.
Go to :
 XML Download
XML Download