

 PDF
PDF Citation
Citation Print
Print



Go to : 
INTRODUCTION
Thrombotic microangiopathy (TMA) is a clinical syndrome characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and subsequent organ damage. Microvascular damage with thrombosis at the arteriolar and capillary level is a common pathological finding that arises from a variety of causes.
TMA syndromes can be classified according to the pathologic mechanism involved as follows: (1) thrombotic thrombocytopenic purpura (TTP), which is caused by a deficiency or decreased activity in ADAMTS13, the von Willebrand factor-cleaving protease, due to genetic mutations or autoimmune inhibitors; (2) typical hemolytic uremic syndrome, caused by Shiga toxin-producing Escherichia coli (STEC-HUS); (3) atypical HUS (aHUS) caused by excessive activation of the alternative complement system upon a genetic predisposition; and (4) secondary TMA, with causes including drugs, pregnancy, hypertension, infection, malignancy, and kidney transplantation (KT).
Accumulating evidence suggests that a large percentage of the patients diagnosed with secondary TMA exhibit genetic abnormalities associated with complement dysregulation. For example, a subset of patients with hypertension-associated TMA exhibits the features of complement-mediated TMA, with a poor prognosis [1]. Genetic abnormalities in genes such as complement factor H (CFH) or complement factor I (CFI) have been found in 29% of patients with de novo TMA after KT [2]. In addition, it has been reported that a substantial proportion of patients with STEC-HUS, pregnancy-associated HUS, or autoimmune TMA have complement variant genes [3,4]. However, more than 20% of patients with aHUS have an unclear genetic background, and both a genetic predisposition and triggering factors are involved in complement overactivation in patients with aHUS [5,6].
Therefore, aHUS and secondary TMA/HUS, which are classified as separate diseases, can be regarded as overlapping diseases that depend on complement dysregulation. This overlapping representation complicates the differential diagnosis of the underlying etiology despite advances in our understanding of the mechanisms of different types of TMA syndrome.
A consensus report was recently published by a group of Korean experts who shared updated opinions on the diagnosis and management of TMA syndrome [7]. They presented updated TMA diagnostic criteria and triggers based on the latest findings, and summarized the treatment strategies for aHUS. In particular, that report emphasized the importance of considering the possibility of aHUS, even in patients with TMA with secondary causes, due to the incomplete genetic penetrance of aHUS.
KT is a representative etiology related to various risk factors involved in the pathophysiology of TMA. Numerous triggers, including cold and warm ischemia-reperfusion injury (IRI), immunosuppressive drugs (such as calcineurin inhibitors [CNIs]), infection, and antibody-mediated rejection, can activate the complement system and cause endothelial damage, regardless of the genetic burden.
Posttransplant TMA is not a rare complication, occurring in 0.8%–29.4% of KT cases, with de novo TMA being more frequent than recurrent TMA/aHUS [8-10]. However, it has been reported that genetic mutations in complement factors are present in approximately 30% of kidney transplant recipients with de novo HUS [11]. The prognosis for overall graft function in TMA is quite poor, especially in recurrent aHUS with a genetic predisposition, and graft failure rates of approximately 40% within 2 years have been reported [9]. Given that TMA has a poor prognosis, KT requires a sacrifice from the donor, and KT is a treatment that places a substantial burden on medical staff and patients, prompt substantial burden for risk factors, including a possible genetic burden, and treatment to ameliorate abnormal complement activation are particularly important.
Here, we review the current understanding of post-KT TMA, as well as various aspects of its pathophysiology and management.
Go to :
Case
In December 2014, a 29-year-old man was admitted for allograft deceased-donor KT. Although he had already undergone allograft living-donor KT from his mother in 2002 because of end-stage kidney disease (ESKD) caused by membranous proliferative glomerulonephritis (MPGN), acute rejection with TMA occurred in the first year after KT. He returned to hemodialysis in the second year, waiting for deceased-donor KT. The human leukocyte antigen and ABO types fully matched the donor. The complement-dependent cytotoxicity crossmatch was negative and there were no donor-specific antibodies (DSAs). The patient underwent immediate KT. After the second KT, the patient’s hemoglobin level gradually decreased to 7.3 g/dL. Moreover, thrombocytopenia (5.6 × 104/μL) developed, the lactate dehydrogenase (LDH) level increased to 953 IU/L, and schistocytes were seen in a peripheral blood smear. Because the improvement in creatinine levels was delayed, a kidney biopsy was performed on the 19th day after surgery. The biopsy findings were consistent with TMA, and acute active antibody-mediated rejection (ABMR) was suspected. To determine the cause of the recurrent TMA, ADAMTS13 activity was evaluated and found to be within the normal range (80%). Genetic testing revealed that the patient was heterozygous for two mutations (c.3572C>T [p.Ser1191Leu] and c.3590T>C [p.Val1197Ala]) in CFH. As eculizumab, a terminal complement (C5) inhibitor, was not available in Korea at the time, plasmapheresis was performed repeatedly starting on the fifth day after surgery, and renal function partially improved. However, the patient was hospitalized several times because of recurrent acute kidney injury (AKI), and despite several rounds of plasma exchange therapy, dialysis was started again 1 year later as renal function gradually deteriorated.
Go to :
GENETIC ABNORMALITIES IN RECURRENT ATYPICAL HEMOLYTIC UREMIC SYNDROME AFTER KIDNEY TRANSPLANTATION
Recent studies on the genetic background of aHUS have revealed significant genetic heterogeneity in several genes involved in complement regulation, including the CFH, CFH-related protein (CFHR1-4), CFI, complement factor B (CFB), complement component 3 (C3), and CD46 (membrane cofactor protein, MCP) genes in the alternative complement pathway [3,12]. However, the factors that induce TMA after KT are diverse, and it remains difficult to identify the genetic predisposition related to complement activation in the transplant environment.
Recurrence risk is determined by genetic mutations in complement proteins. Patients with mutations in CFH, CFH/CFHR1 hybrids, and THBD have a high risk of disease recurrence. Patients with CFI mutations, C3 mutations, anti-CFH antibodies and a negative genetic study are at moderate risk of recurrence, while patients with isolated MCP/DGKE variants are reported to be at low risk of aHUS recurrence after KT [13-15].
It is important to note that the risk of recurrence can also be influenced by other factors such as age, history of KT, and the immunosuppressive regimen. Even in the presence of genetic mutations, various second hits are involved in initiating dysregulation of the alternative complement pathway and promoting endothelial damage and platelet aggregation. Therefore, a multidisciplinary approach is needed to assess risks and develop a management plan for KT recipients with aHUS.
In our patient, IRI, CNI use, antibody-mediated rejection, and genetic CFH defects may have contributed to uncontrolled activation of the alternative complement pathway. In addition, MPGN was the underlying cause of ESKD. Thus, an important aspect to consider is the potential involvement of complement pathway dysregulation in the pathogenesis of MPGN. Immunoglobulin-negative MPGN has recently been classified as C3 glomerulopathy, and like aHUS, dysregulation of the alternative complement pathway resulting from either acquired or genetic causes may contribute to the underlying mechanism. Therefore, in cases where MPGN is identified as the causative disease, it is crucial to re-evaluate the biopsy results and assess the patient for potential predisposition to alternative complement pathway activation before transplantation.
The prognosis of aHUS with genetic abnormalities is generally poor, and a rapid decline in renal function often progresses to ESKD. Disease recurrence is associated with graft loss, and patients with moderate-to-high risk of recurrence and CFH or gain of function (C3, CFB) mutations have the highest risk of graft failure [13,14].
Treatment of recurrent aHUS with a genetic predisposition typically involves the use of drugs that inhibit the complement system, such as eculizumab. Eculizumab improves hematologic and renal outcomes in KT recipients, even in patients with a history of multiple graft losses [16]. Plasma exchange can also help ameliorate hematological abnormalities, such as MAHA, thrombocytopenia, and elevated LDH, while providing healthy complement regulators; however, it has limitations in reversing endothelial damage and end-organ dysfunction. In our patient, TMA occurred twice after KT, and a mutation in CFH led to repeated uncontrolled complement activation with posttransplant trigger factors despite multiple plasmaphereses, resulting in renal damage and graft failure.
Given the incomplete genetic penetrance of aHUS and the fact that both genetic and environmental factors can act together in uncontrolled complement activation, it is important to evaluate the possibility of a genetic predisposition when the treatment response is poor, even when secondary triggers are evident. In addition, prompt and aggressive complement-targeting treatment is required to prevent further damage to transplanted kidneys. In particular, early diagnosis and management are critical to ensure the best possible outcomes for these patients.
Go to :
ENVIRONMENTAL FACTORS IN DE NOVO HEMOLYTIC UREMIC SYNDROME AFTER KIDNEY TRANSPLANTATION
KT promotes several environmental triggers that can induce the excessive activation of alternative complement pathways, including immunosuppressive drugs (e.g., CNI), infections, IRI, surgery, and rejection. IRI alone can activate the complement system by releasing danger-associated molecular patterns, antigen presentation, and sterile inflammation in the kidneys. In an animal model of renal IRI, factor B-deficient mice showed a significant reduction in IRI-induced renal damage, suggesting that activation of the alternative pathway plays an important role in this process [17].
A human study also showed higher expression of genes coding for complement proteins (C1q, C1s, C1r, C2, C3, C4, CFB, and CR1) in kidneys from deceased donors than in kidneys from living donors. Overexpression of complement components is associated with prolonged cold ischemia, indicating that IRI is an important mechanism of complement activation [18]. A recent multicenter study demonstrated that transplants from deceased donors were more closely associated with TMA and that longer cold ischemia time was an independent risk factor for de novo TMA [19].
CNIs increase the incidence of de novo TMA (4%–15%) in KT recipients, and TMA usually occurs early during treatment with high-dose immunosuppressive agents [20]. Cyclosporine has been reported to be more closely associated with TMA than tacrolimus, and several mechanisms have been proposed. The potential mechanisms of cyclosporine- or tacrolimus-induced TMA include alterations in prostacyclin synthesis, loss of equilibrium between vasoactive peptides, and increased shear stress in the vasculature [9,21,22]. Cyclosporine also increases the risk of thrombosis by downregulating the protein C anticoagulant pathway and reducing thrombomodulin activity in endothelial cells [23]. A recent report demonstrated that endothelial cells exposed to cyclosporine in vitro and in vivo released microparticles that activated alternative complement pathways [24].
Although treatment guidelines are not well defined, temporary CNI discontinuation with plasma exchange is widely used for de novo TMA treatment [25]. The most commonly used immunosuppressive strategy for CNI discontinuation is switching to mammalian target of rapamycin (mTOR) inhibitors; however, these drug alteration protocols remain a matter of debate. mTOR inhibitors can also cause TMA, and a recent study found that mTOR inhibitor regimen and recipient age were independently associated with an increased rate of TMA recurrence [9]. Recent reports have stated that the use of belatacept more reliably prevented or treated TMA caused by CNIs [26-28]. However, the effectiveness and safety of conversion protocols using belatacept require further evaluation in larger patient groups.
DSAs can bind to endothelial leukocyte antigens and activate both classical and alternative complement pathways through C1q, C3, and C4 activation [29], resulting in endothelial damage. Therefore, de novo TMA is a histological finding of ABMR that negatively impacts graft survival. Recent reports have shown that allosensitization plays an important role in the development of de novo TMA and increases the risk of early graft loss when TMA is concurrent with ABMR [19,30]. Although DSAs are a strong driver of TMA, the implications of dysregulated complement overactivation may be underestimated in cases of ABMR representing TMA. It is challenging to assess the actual effect of ABMR on the development of de novo TMA and determine the extent to which DSAs alone or uncontrolled complement activation with a genetic predisposition contributes to TMA development. A histological examination revealed that microvascular inflammation, such as glomerulitis and inflammation of the peritubular capillaries, was significantly more prominent in ABMR-related TMA, whereas arteriolar hyaline was predominantly observed in other types of TMA [31,32]. Our case showed the typical microvascular inflammation of ABMR with TMA, but a history of recurrent TMA (first and second KTs) strongly suggested a genetic predisposition, and a CFH mutation was found. Given the poor prognosis of ABMR presenting with TMA, the differential diagnosis and rapid decision-making based on the ABMR treatment response are important. Therefore, complement-targeting drugs may be required for graft rescue in patients with uncontrolled complement activation.
Eculizumab, a terminal complement (C5) inhibitor, was recently shown to be effective in acute ABMR; however, long-term treatment failed to prevent the development of chronic ABMR [33,34]. Another agent that modulates complement activation, a purified C1 esterase inhibitor (C1-INH), significantly improved renal function in patients with acute ABMR in a small-scale randomized controlled trial [35]. A large multicenter study is currently underway to evaluate the effects of C1-INH.
Go to :
EVALUATION, PREVENTION, AND TREATMENT OF POST-KIDNEY TRANSPLANTATION THROMBOTIC MICROANGIOPATHY
TMA should be considered in all patients with AKI, thrombocytopenia, or anemia. If TMA is suspected, a kidney biopsy may be useful for confirming the diagnosis. However, it is important to note that the typical histologic findings associated with TMA, such as arteriolar or capillary thickening, endothelial edema or detachment, and fibrin or platelet-rich thrombi, are nonspecific. Therefore, a biopsy should not delay diagnosis and treatment. When TMA is present, a thorough investigation of hematological abnormalities and secondary causes should be conducted to guide the diagnosis and treatment.
A distinction between STEC-HUS, TTP, and aHUS must be made using an immediate peripheral blood smear, testing for ADMTS13 activity, and Shiga toxin assays. If TTP and STEC-HUS are excluded, the possibility of aHUS and early treatment with plasma exchange and eculizumab should be considered. Importantly, the absence of a detectable genetic abnormality and the presence of a TMA-related condition did not preclude a diagnosis of aHUS.
Therefore, if TMA of unknown cause occurs after KT or if recurrent TMA is observed, aHUS should be differentiated immediately. In addition, since there are numerous triggering conditions after KT, if secondary TMA is suspected, treatment for the inducing factors should be performed first; however, if there is no response to treatment for secondary causes or plasmapheresis-dependent patterns are noted, an inherent deficiency of complement-regulating proteins should be considered (Fig. 1). Transplant outcomes in patients with aHUS are particularly poor, with graft loss rates of approximately 24% at 1 year and 49% at 5 years after KT [13].
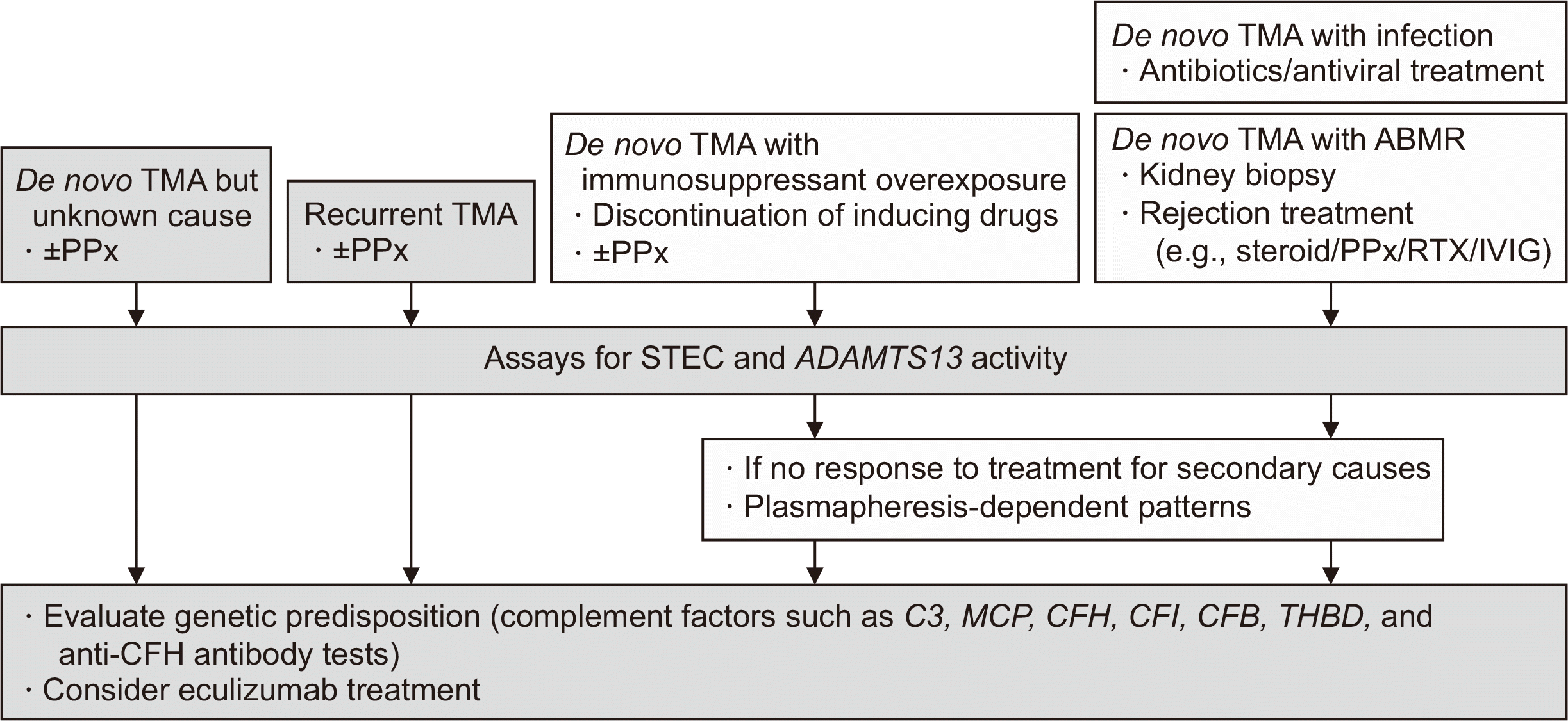 | Fig. 1Diagnostic algorithm and therapeutic options for posttransplant TMA. If TMA of unknown cause occurs after kidney transplantation or recurrent TMA is observed, ADAMTS13 and Shiga toxin assay tests need to be performed to differentiate aHUS from TTP and STEC-HUS, along with genetic testing, and consider early initiation of eculizumab. If secondary TMA is suspected, trigger management should be performed first, but if there is no response to treatment for the secondary cause or if plasmapheresis-dependent patterns are observed, the diagnosis and treatment of aHUS should be considered. TMA, thrombotic microangiopathy; PPx, plasma exchange; ABMR, antibody-mediated rejection; RTX, rituximab; IVIG, intravenous immune globulin; STEC, Shiga toxin-producing Escherichia coli; MCP, membrane cofactor protein; CFH, complement factor H; CFI, complement factor I; CFB, complement factor B; THBD, thrombomodulin; TTP, thrombotic thrombocytopenic purpura; HUS, hemolytic uremic syndrome; aHUS, atypical HUS.
|
Plasma exchange or plasma therapy usually supplies normal complement regulatory proteins and can correct hematologic abnormalities, but it does not improve graft function and is limited in improving patient outcomes [25]. However, since the introduction of eculizumab, the outcomes of aHUS transplant patients have improved compared to those of patients with aHUS who are not treated with eculizumab [16,36]. In particular, the early initiation of eculizumab after development of the clinical features of TMA is associated with better recovery of renal function [37], and in patients with genetic variants of the complement system, the prophylactic use of eculizumab was independently associated with a significantly reduced risk of recurrence and longer graft survival [38]. A recent study compared transplant outcomes between the prophylactic use of eculizumab at or prior to KT and rescue treatment after KT and demonstrated better 2-year renal function and graft survival in the prophylactic treatment group than in the rescue treatment group, indicating that a pre-KT evaluation for genetic predisposition to aHUS is important in cases of recurrent TMA or unknown ESKD [36]. An expert recommendation is that patients with aHUS at high or intermediate risk of recurrence should receive prophylactic eculizumab treatment, and patients at low risk should be informed of the risk of recurrence and closely monitored after KT [12].
The decision to pursue a third KT for our patient, who has experienced two prior failed transplants due to TMA recurrence, is a complex process that would necessitate a careful consideration of various factors, such as the patient's overall health status, comorbidities, and the availability of suitable donor organs. Despite the high risk of aHUS recurrence in patients with CFH mutations, prophylactic administration of eculizumab has been demonstrated to effectively prevent aHUS recurrence in a significant proportion of patients. Additionally, since CFH is primarily produced in the liver, liver transplantation could be an alternative treatment option [39,40].
In Korea, eculizumab has been covered by national insurance since 2018 as a treatment for aHUS. However, approval from the Health Insurance Review and Assessment Institute is required before it can be administered to a patient. Therefore, it is difficult to start eculizumab early because of the review process, and prophylactic treatments to prevent recurrent TMA in patients with aHUS have not yet been approved. Considering the special circumstances of transplantation, such as single kidney function, donor sacrifice, and the poor outcomes of TMA, efforts to broaden the treatment indications for patients with transplanted kidneys are needed.
Patients with a genetic predisposition may require lifelong eculizumab treatment. However, if there is no recurrence for several months after at least 6–12 months of treatment, discontinuation can be considered on a case-by-case basis. No clear guidelines on the criteria for discontinuing eculizumab therapy in patients with aHUS are available to date, and systematic studies are needed to determine these criteria.
Recently, new complement-targeting drugs such as long-acting C5 inhibitors, oral drugs, and C5a receptor antagonists have been developed, and studies have also investigated the efficacy of C5 inhibition in complement-related diseases, including aHUS [41].
Go to :
CONCLUSIONS
TMA is not a rare complication of KT; therefore, it should be promptly diagnosed and treated. Because there are multiple triggers for TMA in the context of KT and the gene expression in aHUS is incomplete and often occurs in response to triggers, secondary TMA and aHUS are considered clinically overlapping disease entities and are difficult to differentiate clinically. Since TMA that occurs after KT has a very poor prognosis, a rapid diagnostic process, trigger management with or without plasmapheresis, and early administration of eculizumab when aHUS is suspected are important. Considering the high cost of eculizumab treatment, attention to and improvements in insurance policies and national support are needed. Further studies on comprehensive treatment indications in patients, including those with secondary TMA and aHUS, are needed to elucidate the effects of new complement-targeting drugs.
Go to :
 XML Download
XML Download