

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Coffin-Siris syndrome (CSS, MIM 135900) is a disorder characterized by distal phalanx, nail hypoplasia, or aplasia of the fifth finger or toe (digit), which may be accompanied by developmental or cognitive delay, characteristic facial features, hypotonia, hypertrichosis, microcephaly, and organ system abnormalities [1, 2]. The syndrome is caused by variants in several genes which are encoding components of Brahma-related gene 1 (BRG1)/ Brahma homologue (BRM)-Associated Factor (BAF) complex [3], which is one of the chromatin-remodeling complex families known in mammals [4, 5]. These genes include ARID1B, ARID1A, SMARCB1, SMARCA4, SMARCE1, ARID2, DPF2, SMARCC2, SOX11, SOX4, SMARCD1, and BICRA, each corresponding to CSS subtype 1-12, respectively [1, 2, 6]. Although phenotypic features for each CSS subtype have been established in previous studies [3], CSS cases without finger- or toe-related findings, which are representative phenotypes of some CSS subtypes, have also been reported recently [2].
A genotype-phenotype correlation study is an effective way to relate individual genetic backgrounds to specific diseases or characteristics, and is the basis of precision and genomic medicine [7]. The purpose of this study was to analyze genotype-phenotype correlations in patients with suspected developmental delay (DD) and intellectual disability (ID), and clinically suspected CSS, and to identify novel variants. This study was approved by the Institutional Review Board (IRB) of Seoul National University Hospital (IRB No. 2204-161-1320) under the Helsinki Declaration.
MATERIALS AND METHODS
This retrospective study reviewed the electronic medical records of 23 suspected patients with CSS enrolled in a rare disease diagnostic program of the Korean Disease Control and Prevention Agency (KDCA) from January 2017 to December 2020. Patients were enrolled when they exhibited the following signs or symptoms, and demonstrated potential clinically suspected CSS: dysplasia of either the distal phalanx or the nail of the fifth digit, DD, ID, coarse facial features, hypertrichosis, seizures, and central nervous system (CNS) malformations. Among the 23 patients with suspected CSS, who requested for a rare disease diagnostic program, eight cases reported positive for CSS were included, and the remaining patients were excluded. For the CSS positive patients, basic information (reason for test, test date, diagnosis, age at diagnosis, sex), clinical manifestations (such as finger or toe hypoplasia, DD/ID, characteristic facial findings, hypotonia, scalp hair condition, cardiac anomaly, gastrointestinal tract anomaly, and CNS anomaly), and laboratory test results (reports from rare disease diagnostic program) were collected retrospectively from electronic medical records.
In the rare disease diagnosis program, whole-exome sequencing (WES), Sanger sequencing and multiplex ligation dependent probe amplification was performed. Then, variants were classified based on the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines [8].
The R version 4.2.0 (R Core Team, Vienna, Austria) was used for statistical analysis. The frequency and ratio of binary phenotypes per gene were calculated and visualized. A cluster analysis using Jaccard/Tanimoto similarity test [9] was performed to confirm if the cluster was formed according to genotype-phenotype, and the significance of the clusters. The Jaccard/Tanimoto coefficient is the ratio of union to intersection and is used to measure the similarity between binary data [9]. The network between clusters was expressed as a graph using the calculated P-value. The Leiden Open Variant Database [10], ClinVar, and literature review were used to determine if the pathogenic variants identified in this study have been previously reported.
RESULTS
The frequency of variant types and phenotypes for each causative gene is summarized in Table 1. In ARID1B, four cases of nonsense and each case of frameshift and splicing were identified, and two cases of missense were identified in SMARCA4 (Table 1). In the cases of ARID1B, CNS anomaly and DD/ID were observed in all cases, and hypertrichosis was observed in five cases. Among the CNS malformations, seizures were the most common in three cases, followed by shortening of corpus callosum in the two cases. In the cases of SMARCA4, hypoplasia of the fifth digit and unique facial features were observed in all cases (Table 1).
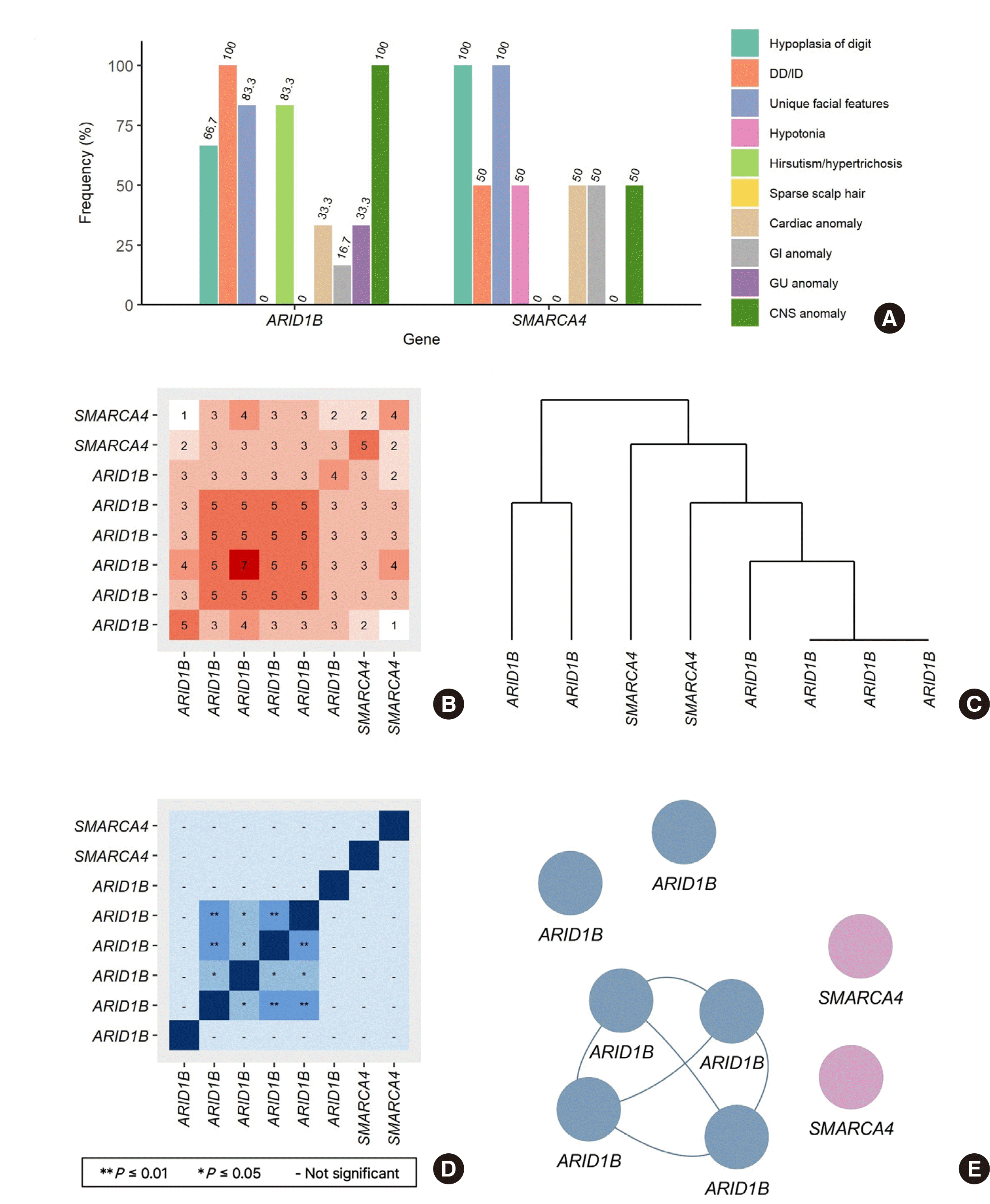
In Fig. 1A, the frequency of phenotypes observed by each gene is expressed as a bar plot. In the combination of cases, the number of phenotypes shared by the two cases was expressed as a matrix (Fig. 1B), and five phenotypes were observed identically in three cases of ARID1B. In a dendrogram based on the matrix (Fig. 1C), it was also confirmed that three of the ARID1B cases had the same phenotype completely, but some ARID1B cases showed more similar phenotypes to that of SMARCA4 cases. The adjacency matrix (Fig. 1D) was calculated using the Jaccard/Tanimoto similarity test of phenotype matrix (Fig. 1B), and case pairs showing significant similarity were indicated. A network with significant similarities between each case is graphically represented in Fig. 1E. The edge-connected nodes indicate a significant similarity. These nodes had common phenotypes: hypoplasia of the fifth digit, DD/ID, unique facial features, hypertrichosis, and CNS anomaly.
To the best of our knowledge, five novel variants were identified in this study (Table 2). In ARID1B cases constituting the genotype-phenotype cluster, three of the four cases showed novel variants (Gly1696*, Gly806Trpfs*, and Asn1320*). The novel variant (Asn916Ser) of SMARCA4 was a de novo variant identified through a trio test.
DISCUSSION
In this study, we confirmed that a subgroup of ARID1B has a significant similarity in terms of phenotype. In addition, to date, we have identified five new variants of CSS-causative genes including a de novo gene that was not previously reported to the best of our knowledge.
CSS is a genetically heterogeneous disease, and it has been previously established that the variants of ARID1B among the causative genes account for the largest proportion (up to 83%) of CSS [3, 11]. Although only two causative genes, ARID1B and SMARCA4, were identified in this study, the proportion of causative genes is congruous with previous studies [11]. Several ARID1B-associated CSS patients were previously reported to have various levels of DD/ID, hypertrichosis, and unique facial features such as low anterior hairline and thick eyebrows, however, hypoplasia of the fifth digit was observed in some ARID1B cases only [3, 11]. Patients with CSS having pathogenic variants of ARID1B are known to have relatively mild symptoms and normal growth [11]. ARID1B is one of the subunits that constitutes BAF complex, and is known to play a role in regulating the function of chromatin complexes [5]. In this epigenetic mechanism, ARID1B plays an important role in regulating gene activity by inducing the expression of proliferative genes, and promoting the cell cycle involved in neural stem cell differentiation [5, 12]. This could be inferred as the reason for CNS anomaly identified in all ARID1B cases in this study. The occurrence of episodes such as seizures in patients with suspected CSS, indicates the possibility of pathogenic variants in BAF-related genes, which should be confirmed [13]. In our study, the zygosity of every variant in ARID1B included heterozygous truncating variants, thus, it is expected to cause loss of function due to haploinsufficiency followed by the allelic decrease of functional gene product [2, 3, 5]. One splicing variant identified in ARID1B is located at the last base of exon 17, which is expected to cause alternative splicing in-silicon prediction using programs such as NNSPLICE, and GeneSplicer, and was reported to cause exon 17 skipping in a previous functional study [14]. In 40-80% of ARID1B-related diseases, including CSS, hypotonia is known to be present [15]. However, the medical records of ARID1B cases in this study did not contain explicit references to hypotonia. Hypotonia, which can be caused by anomalies in CNS or lower motor neurons, is a sign of decreased tone of the skeletal muscles and decreased resistance to passive stretching [16]. Seizures or global DD/ID including cognitive delay are frequently present in patients with central hypotonia, which has a mild peripheral muscular weakness [16, 17]. Several possibilities can be considered because hypotonia could not be identified in this study. In the case of CSS, hypotonia typically manifests in infancy [11], however, most patients in this study were diagnosed during their childhood or adulthood stage. Therefore, it is difficult to rule out the possibility that the evaluation of hypotonia was omitted during the medical history collection or the physical examination. However, considering most ARID1B cases in this study were also accompanied by delayed motor and cognitive development, and that DD was verified in four of the six cases, the likelihood of hypotonia cannot be fully ruled out. To unequivocally confirm this, a clinical re-evaluation of the patients with the ARID1B variants may be required.
SMARCA4 accounts for about 7% of CSS causes, which is the second-highest proportion [11]. In previous studies, it was established that patients with CSS with SMARCA4 variants almost always have hypoplasia of the fifth digit, many of them have hypotonia and behavioral abnormalities, and half of them have DD/ID [3, 11]. SMARCA4 is also a component of the BAF-complex, which forms a central ATPase. Heterozygous missense variants in SMARCA4A have a dominant-negative effect rather than haploinsufficiency, altering chromatin accessibility and making it difficult to bind to the super-enhancer [2, 18]. All our SMARCA4 cases were identified as heterozygous missense, all of them showed hypoplasia of the fifth digit, and half of them showed DD and hypotonia. These are consistent with the results from previous research [2, 11, 18]. In the case of a confirmed de novo variant of SMARCA4 Asn-916Ser, the patient’s older brother, sister, and parents were not affected, and it was confirmed that they did not have the variant in the trio test. The following symptoms were identified in this particular patient: a dysmorphic face, dysplasia of corpus callosum, dysmorphism of the second toe, and patent ductus arteriosus. Short stature, generalized laxity and hypotonia were presented as musculoskeletal manifestations. Generally, de novo variants are more harmful than other genetic variants inherited because it has been subjected to relatively less stringent evolutionary selection [19]. De novo variants are known to be a major cause of early onset genetic disorders such as Kabuki and CSS [19]. De novo variants in each BAF-related genes (ARID1A, ARID1B, SMARCB1, SMARCA4, SMARCA2, and SMARCE1) were reported to cause CSS, suggesting that abnormalities of BAF-complex cause abnormal neurodevelopment of CSS [20]. In addition, the variant is located in the ATP-ase domain of SMARCA4, and the site is also a mutational hotspot [21]. Missense variants in the SMARCA4 ATPase domain impair the regulatory mechanism of PRC1 protein related to methylation, thereby reducing the chromatin remodeling activity of the BAF-complex [21, 22].
There are some caveats in interpreting the results of this study. First, there may be limitations in generalization because the sample size is too small and patients were tested according to their clinical manifestation; one alternative solution is to analyze multi-omics together with genomic information. Second, a cluster with similar phenotypic networks was identified in a subset of specific causative genes, but owing to the nature of the retrospective study, CSS phenotypes that are not mentioned in the electronic medical record may exist, which may lead to changes in phenotypic networks and consequently, new clusters may emerge or existing clusters may disappear. Thus, it is expected that the cases that formed a single-member cluster in this study could be reclassified according to additional phenotypic information.
In conclusion, we reported four patients with a novel ARID1B variant and one patient with a novel SMARCA4 variant which is de novo. We also identified that there was a significant genotype-phenotype correlation between cases. This study contributes to describing the CSS phenotype caused by ARID1B or SMARCA4 variant. Future studies may facilitate easier diagnosis of CSS in patients who present with atypical traits using more in-depth genetic testing, such as WES, when applied to previously poorly known rare disorders. Molecular genetic testing may lead to further research on the genetic diagnosis, clinical symptoms, classification of this rare disease and criteria.
 XML Download
XML Download