

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
There are many genetic disorders or syndromes that cause intellectual disabilities, and they are usually accompanied by inconsistent symptoms or abnormalities. OPHN1, one of the causative genes of hereditary intellectual disability, is located in Xq12 and encodes an oligophrenin-1 protein that plays an important regulatory role in the development and migration of fetal nervous tissues [1]. As we are aware, from 1997 to the present, there have been several case reports of wide spectrums of X-linked intellectual disabilities (XLIDs) associated with variants of the OPHN1 gene. [2–9]. XLIDs caused by OPHN1 gene mutations have mental retardation and cerebellar hypoplasia in common, and other features such as epilepsy, ataxia, strabismus, urogenital abnormality, facial dysmorphism, and ventriculomegaly can also be observed. In this case study, we report two cases of OPHN1-associated XLID found in one family, the first to our knowledge, in Korea. The requirement for informed consent was waived (IRB No. 2022-05-060-001) by Institutional Review Board of Keimyung university Dongsan Hospital.
CASE
In 2015, a 23-month-old boy visited the rehabilitation department with a general developmental delay. He was his parent’s first child and was born at full term following a normal pregnancy via vaginal delivery; his body weight was 3,300 g. He presented with facial dysmorphisms, including a prominent forehead, ocular anomalies (bilateral ptosis, horizontal nystagmus, and impaired visual perception), a prominent nose with a wide nasal base, a high palate, dental cavities, and pes planus. A prominent cystic lesion in the retrocerebellar convexity suggesting mega cisterna magna findings was observed on the brain MRI; the laboratory tests showed no abnormal results with the 46,XY karyotyping. During his neurological examination when he was 7 years old, he exhibited poor fine motor coordination and the inability to organize activities of daily living. The psychological examination showed moderate intellectual disability with a low spatial orientation, a working memory with short-term memory capacity, emotional instability, and poor concentration. The personal and social adaptive functions were equivalent to that of a child aged 53 months (SQ=56.19). The receptive language age was 47 months and the expressive language age was 41 months.
In 2021, his younger brother, a 45-month-old male, visited the rehabilitation department with a developmental delay as well as a retro-cerebellar cystic mass and diffuse frontal cortical atrophy from birth. He was born at intrauterine pregnancy of 38 weeks with a body weight of 2,600 g. During the clinical examination, we observed the following characteristics: discrete facial dysmorphisms, pes planus, and functional scoliosis with poor posture. The psychomotor examination revealed delayed gross motor development with poor balance on walking ambulation and was equivalent to that of a child aged 30 months. The personal and social scales were equivalent to a child aged 30 months, and the language scale age was 25 months. He was also taking medicine for hypothyroidism.
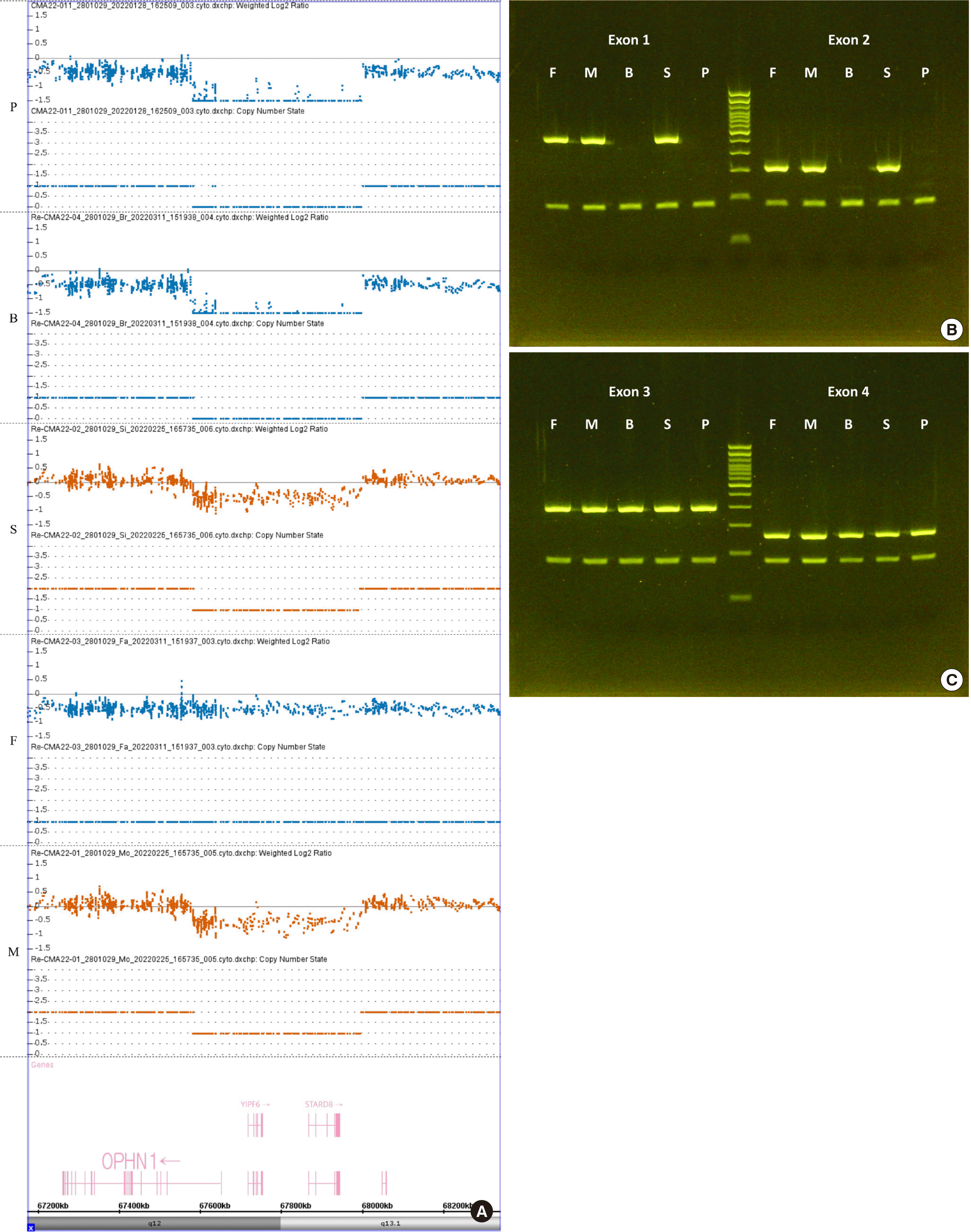
A chromosomal microarray (CMA) test was performed with the GeneChip® System 3000Dx v.2 (Thermo Fisher Scientific, Waltham, MA, US) using a CytoScan™ Dx Assay Kit (Cat. No. 902420, Thermo Fisher Scientific), to confirm the hereditary intellectual developmental disorder. As a result of the CMA test, a hemizygous 360 kbp deletion (chrX: 67,636,890-67,997,055) containing a part of OPHN1, known as the causative gene for XLID, was discovered (Fig. 1A). To confirm the partial deletion of the OPHN1 gene, a PCR test was performed using primer pairs for exons 1, 2, 3, and 4, and the deletion of exons 1 and 2 was confirmed (Fig. 1B, C). The identical hemizygous deletion was found in the proband’s younger brother with similar symptoms. In additional CMA and PCR family tests, the heterozygous deletion was revealed in the unaffected mother and younger sister. The father showed no copy number variation in OPHN1. The primers and targets of the PCR study are described in Table 1.
DISCUSSION
Several types of mutations have been reported for XLIDs related to OPHN1 mutations, from single nucleotide substitutions to exon deletions, and they have different symptoms depending on the location and type of mutation [2]. For this reason, case reports including the exact variant description are important for genotype-phenotype relation studies. The OPHN1 exons 1 and 2 deletion observed in the cases in this study are suspected to have a similar effect to the case reported by Bienvenu et al. [3], which involved a female patient with mental retardation, epilepsy, ventriculomegaly, and strabismus. She had an X;12 balanced translocation and Billuart et al. [10] identified that this translocation happened in the second intron between exons 2 and 3 and revealed no expression of OPHN1 in the patient.
On the other hand, several studies on the physiological function and pathway related to the Rho-kinase activity of the OPHN1 gene have been reported [1, 11]. Recently, Wang et al. [12] investigated the exact mechanism of stress-related behavior commonly found in OPHN1 gene deficiency, and at the same time suggested its potential as a therapeutic target through animal model experiments. As such, in the case of hereditary intellectual disabilities, precise target treatments may appear depending on the results of future research, so it is important to identify the exact trigger gene for each patient and to accumulate accurate knowledge about the genotype-phenotype relationship. However, in many individual hereditary intellectual disabilities, it is difficult to differentiate between them based on their clinical symptoms alone due to inconsistent phenotypes. Therefore, when genetic intellectual disability is suspected due to the presence of family history as in these cases, it is expected that CMA can be used as an important diagnostic test along with other tests such as whole exome sequen cing.
 XML Download
XML Download