

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Variants in SLC29A3 can lead to a spectrum of inherited syndromic histiocytosis that encompasses H syndrome, pigmentary hypertrichosis with insulin-dependent diabetes mellitus syndrome, Faisalabad histiocytosis, and familial Rosai-Dorfman disease [1,2]. Despite the fact that the precise pathogenesis of syndromic histiocytosis is unknown, the SLC29A3 gene, which encodes the human equilibrative nucleoside transporter 3, is mostly found in intracellular structures and is involved in nucleoside transport, as well as plays a key role in nucleotide synthesis and lysosomal integrity by regulating nucleoside trafficking. Thus, a loss-of-function variant in SLC29A3 leads to lysosomal nucleoside buildup, alteration in macrophage function, and disruption of normal histocyte proliferation and function [3,4].
Various SLC29A3 variants have been identified. Nonetheless, patients with SLC29A3 variants typically have hyperpigmentation, hypertrichosis, hepatosplenomegaly, sensorineural hearing loss, diabetes mellitus, and hypogonadism. It should be noted that some patients may exhibit phenotypic intrafamilial variability [1].
In recent years, several novel clinical findings have gained more attention. Among the phenotypic features are joint contractures and arthritis [5-7]. However, a causal relationship between juvenile inflammatory arthritis and genetic defects is rarely considered. Recent research suggests that SLC29A3-related disorders are accompanied by autoinflammation and autoimmunity due to an overactive inflammasome pathway; most likely induced by mitochondrial and lysosomal dysfunction. As a result, these conditions have been classified as monogenic autoinflammatory conditions [8,9]. Here, we report a girl with a novel variant of the SLC29A3 gene who presents with clinical features that mimic systemic juvenile idiopathic arthritis (JIA) and hyperferritinemia.
All data was collected anonymously, and the confidentiality of the patients was protected. Furthermore, informed consent has been obtained from the patient’s parents for the patient’s photography and to access and collect data from the medical record to be used in scientific publications. This data was part of a study approved by the ethics committee of the Research Affairs Council (RAC) at King Faisal Specialist Hospital and Research Center (RAC no. 2221191).
CASE REPORT
Clinical presentation
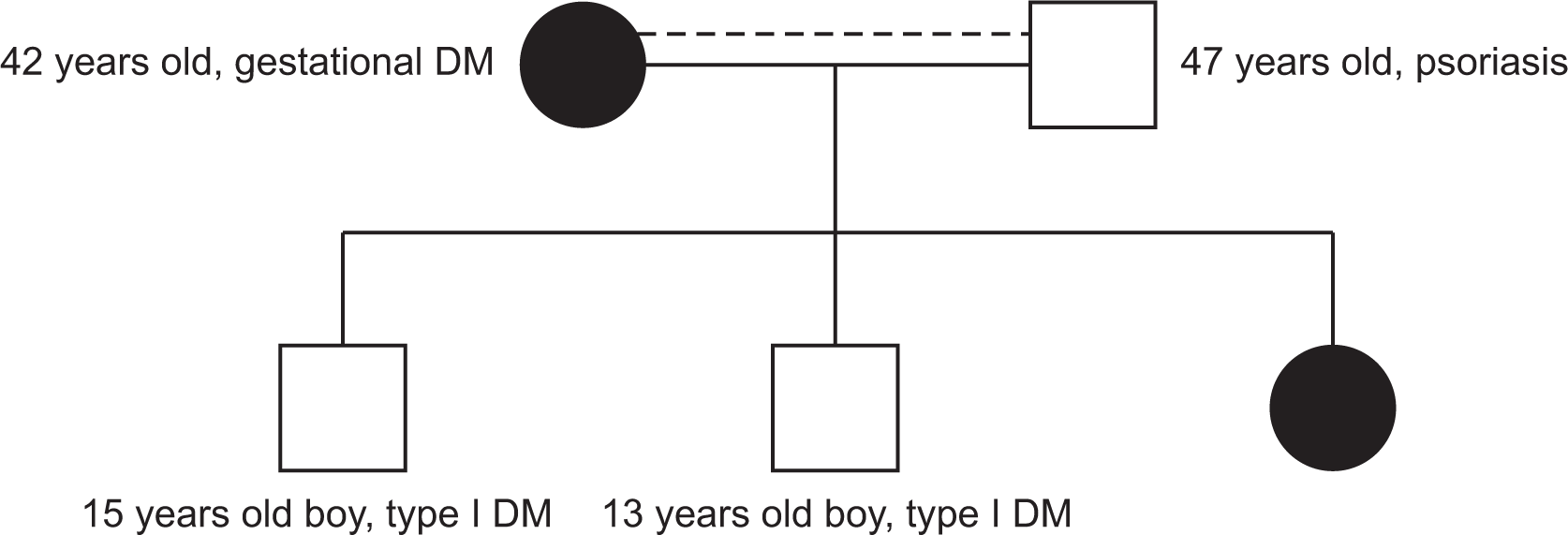
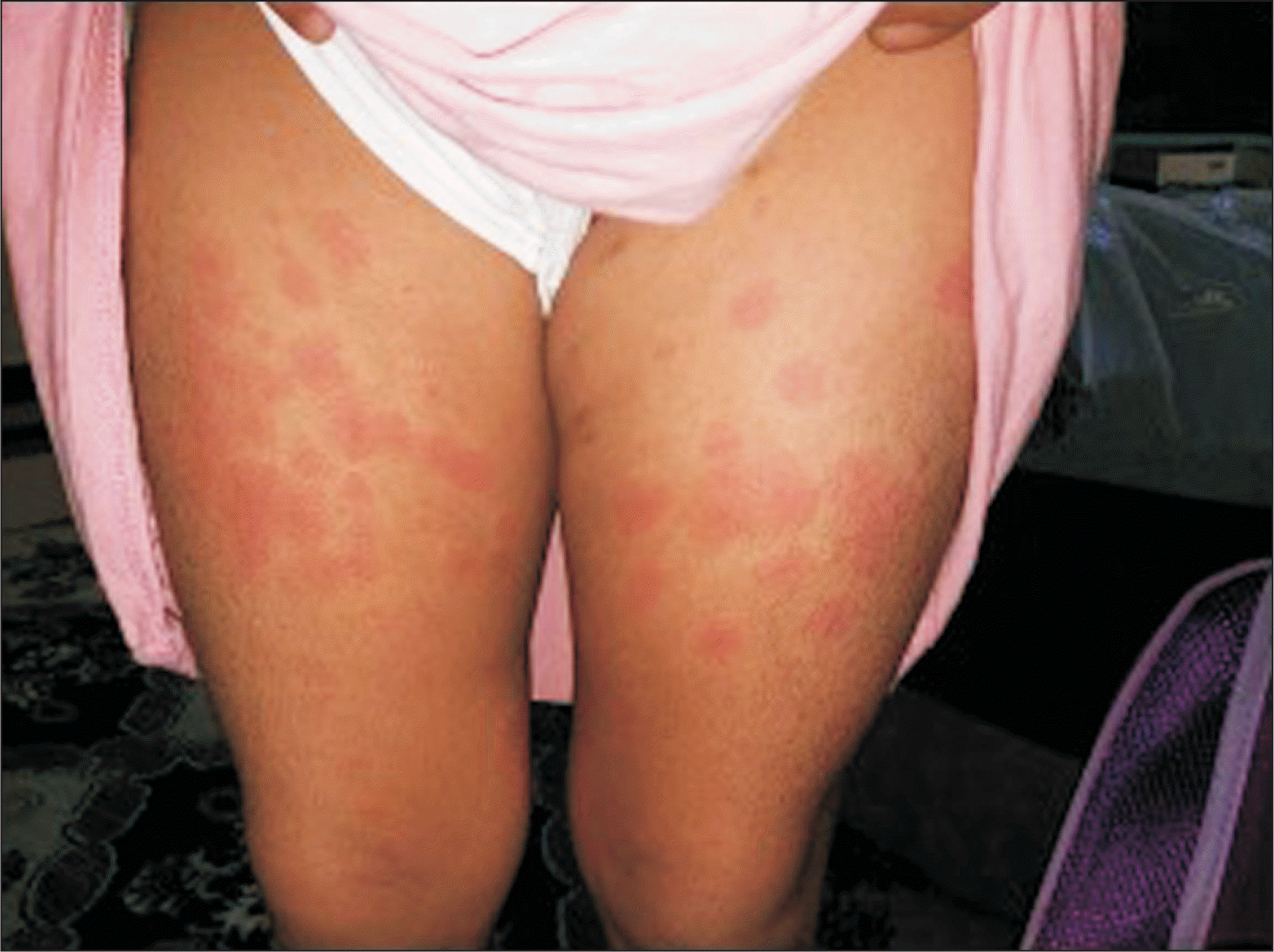
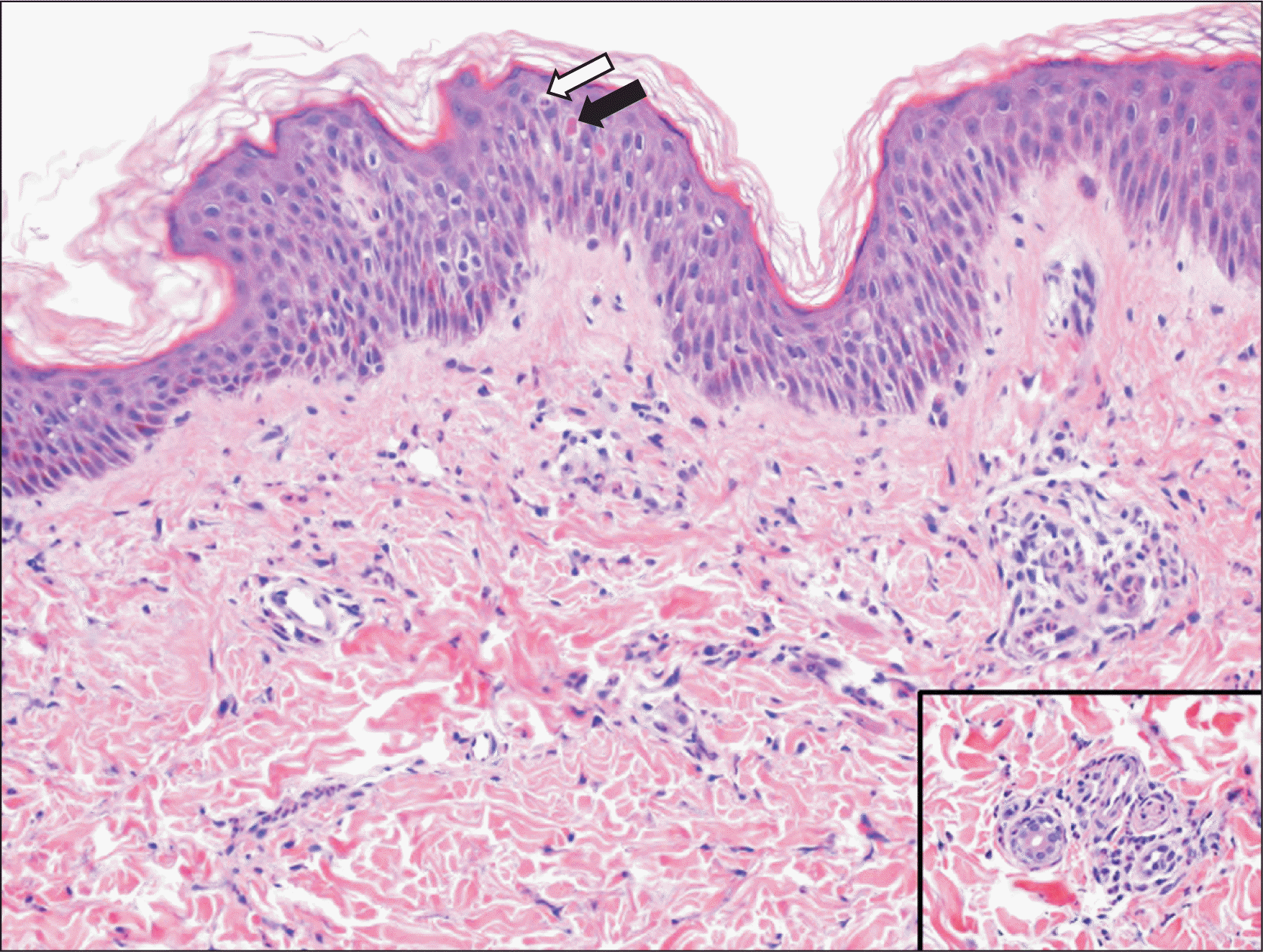
An 8-year-old girl has had hypothyroidism and insulin-dependent diabetes mellitus (IDDM) since she was three years old. She was referred to our pediatric rheumatology service at the age of five years due to a two-month history of quotidian fever, which was associated with a diffuse maculopapular rash and polyarthritis involving the ankles, wrists, and knees. She was appropriately developed for her age, apart from delayed speech abilities. The parents are cousins. The father has psoriasis and the mother has a history of gestational diabetes mellitus, and they have two children with IDDM (Figure 1). Physical examination revealed a subtle dysmorphism in the form of frontal bossing, thick eyebrows, a flat nose, and a long philtrum. Growth parameters were within the 50th percentile, and the tanner stage was appropriate for her age. She had an erythematous maculopapular skin rash over her extremities (Figure 2) and hepatosplenomegaly. There was no hyperpigmentation, hypertrichosis, lymphadenopathy, joint deformity, nor contractures. An ophthalmologic examination revealed prominent eyes with violaceous to dusky red rash on both upper eyelids but no evidence of uveitis. Her laboratory results revealed leukocytosis, thrombocytosis, microcytic hypochromic anemia, and elevated inflammatory markers (erythrocyte sedimentation rate, C-reactive protein), with hyperferritinemia. Table 1 summarized the laboratory findings on initial presentation and last follow-up visit. Bone marrow aspiration showed normocellular bone marrow with active trilineage hematopoiesis and no evidence of abnormal cells. Skin biopsy revealed perivascular and interstitial neutrophilic infiltrates with minimal karyorrhexis and a negative immunofluorescence study (Figure 3). Because of the constellation of the clinical and laboratory findings, she underwent genetic testing. The whole exome sequencing (WES) was ordered. At the time of blood extraction, informed consent for genetic testing was obtained from the parents as part of patient care.
Molecular genetic findings
WES (Baylor Genetics) of the complete coding region of the SLC29A3 gene revealed a homozygous variant in SLC29A3 (NM_018344.5: c.707C>T [p.T236M]) in our patient. This finding is consistent with inherited syndromic histiocytosis, including H syndrome. Both parents were found to be heterozygous for this variant. Both siblings had heterozygous variants, but one of them had a wild type. They are both known cases of IDDM, but had no other features suggesting inherited syndromic histiocytosis. This variant has not been reported in any other patient.
Hearing evaluation and echocardiography were performed as part of comorbidities seen in inherited syndromic histiocytosis and revealed normal findings. Hemoglobin electrophoresis was consistent with the beta-thalassemia trait. A hormonal assay confirmed hypothyroidism; however, gonadal hormone levels were within normal limits.
The patient was labeled as having SLC29A3-associated systemic JIA with hyperferritinemia. Corticosteroids were avoided due to her poorly controlled IDDM. She was managed initially with cyclosporine (4 mg/kg/day) and interleukin (IL)-6 blockade (Tocilizumab, 8 mg/kg, every two weeks); however, it was stopped after the second dose due to a severe infusion reaction. She was then started on IL-1 blockade (Anakinra, 4 mg/kg/day, daily subcutaneous injection) with excellent therapeutic response, her fever and skin rash disappeared, and her leukocytosis, thrombocytosis, and anemia resolved with normalization of inflammatory markers and ferritin levels.
DISCUSSION
The classic clinical presentation of SLC29A3-related syndromes may be accompanied by novel phenotypes. Despite phenotypic variation, the laboratory findings indicate systemic inflammation [8]. Flexion contractures of the fingers and toes have been reported frequently as musculoskeletal manifestations of SLC29A3-related syndromes. However, there have been more published reports of patients with inflammatory arthritis [5,10,11]. Here, we describe a patient who was initially labeled as having systemic JIA based on the International League of Associations for Rheumatology classification criteria for JIA [12]. However, due to the presence of other clinical manifestations, namely IDDM and a novel homozygous SLC29A3 variant, it was determined that the diagnosis was consistent. In addition, there was parental consanguinity and a family history of IDDM and heterozygous SLC29A3 variants. In contrast, our patient did not exhibit many of the same manifestations as those reported in literature, specifically, hyperpigmentation and hypertrichosis. Furthermore, the histologic findings revealed neutrophilic-rich inflammatory dermatoses, which had not previously been reported in SLC29A3-related syndromes. These histologic findings are non-specific, though, and may be present in Still's disease; other histologic considerations include Sweet syndrome and Sweet-like neutrophilic dermatosis. Given the complexity of this case, questions remain unanswered: Could she develop more symptoms and evolve into a more distinct phenotype? Is there a new phenotype associated with this gene variant? Could these constellations be a result of chance? While any of these explanations could be true, we have a tendency to believe that the SLC29A3 variant in this case has caused a novel phenotype with an autoinflammatory nature, particularly with relatively recent reports of novel features in patients with SLC29A3-related syndromes. It is worth noting that, regardless of the underlying mechanisms, it is widely accepted that many autoinflammatory and autoimmune manifestations are linked to immunedysregulation status. Currently, besides the classic phenotype, molecular genetic analysis and histological features of the cutaneous lesions seem to be the most useful for diagnosis. However, some patients with SLC29A3 variants had unique clinical phenotypes and atypical histopathology. This could be due to a remarkable genetic mechanism that prevents the expression of normally coding transcripts [13].
Control of the ongoing inflammatory process and achieving remission are the target of treatment for SLC29A23-related diseases. As a result, although with limited clinical and laboratory improvement, conventional disease-modifying antirheumatic medications such methotrexate, azathioprine, and cyclosporine are frequently utilized. Biologic treatment is rarely used in these rare entities, and its use is limited to anecdotal reports. In a few patients, treatment with IL-6 blockade exhibited improved inflammation [5,9,14]. Interestingly, two patients with SLC29A3 and arthritis showed poor response to anti-TNF biologic agents, then switched to IL-6 blockade with significant improvement [5]. Another patient with autoinflammatory features was given a trial of daily IL-1 blockade with no clinical impact on her systemic inflammatory features and markers. It was then switched to IL-6 blockade, which resulted in a markedly successful response [14]. Furthermore, because of the pathogenic similarities with systemic autoinflammatory disorders, we were able to successfully treat our patient with Anakinra. As a result, IL-1 blockade may be considered an off-label treatment option for such patients.
SUMMARY
We present a patient with findings, namely neutrophilic-rich inflammatory dermatoses, which have not been previously reported in patients with the homozygous SLC29A3 variant; hence, should be considered in the differential diagnosis of patients with systemic juvenile arthritis and undifferentiated systemic autoinflammatory disorders. This report, we hope, will broaden the clinical phenotype and therapeutic options. More patients and follow-up are needed to confirm these findings.
 XML Download
XML Download