

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Tissue transplantation is a medical procedure that replaces a damaged or missing organ by removal of a tissue, or a part of it, from another person (donor) and placing this tissue into the body of a recipient [1]. Transplantation is necessary when the recipient's organ fails; this can occur due to a genetic condition, such as polycystic kidney disease, cystic fibrosis, or heart defects, or as a result of infections, such as hepatitis, physical injuries, or due to chronic conditions, such as diabetes. A tissue transplant can prolong a person’s life and allow them to have a normal lifespan despite chronic illness; therefore, tissue transplantation has become a major advancement in novel medicine [2].
Nonetheless, tissue rejection is a major problem in transplantation; specifically, this tissue rejection involves an immune response against the received tissue, which may lead to transplant failure and immediate organ removal from the recipient.
Human leukocyte antigen (HLA) sensitization occurs after most transplantation cases; therefore, HLA class I and II matching is essential for a successful transplant outcome. In addition to the HLA system, minor histocompatibility antigens (MiHA) are receptors on the cellular surface that can cause rejection in organ transplants; nonetheless, this MiHA–driven rejection occurs less frequently than those of the major histocompatibility complex (MHC) [3].
Allogeneic graft rejection, particularly hematopoietic stem cell (HSC) transplantation, can be mediated by host natural killer (NK) cells alongside NK, γδ, CD4+, and CD8+ T-cells that recognize MiHA on donor cells [4]. Establishing this immunological tolerant state before transplant of hematopoietic stem cells can remarkably improve the clinical outcome of this procedure in terms of overall and disease-free survival [5].
Go to : 
GRAFT-VERSUS-HOST DISEASE (GvHD)
GvHD is another complication in organ transplantation, particularly in the transfer of organs that are that contain many immune cells. Under these conditions, the infused donor immune cells react to the histocompatibility differences between themselves and the host. Despite advances in hematopoietic stem cell transplantation (HSCT), including high-resolution HLA genotyping and routine prophylaxis based on calcineurin inhibitors (CNIs), GvHD incidence still occurs in approximately 50% of cases; specifically this process occurs in 30–50% and 70% of recipients allografted with matched-related and matched-unrelated donors, respectively, and is caused by donor T and B lymphocytes that recognize and attack host antigens [6]. During this event, immunologically-competent donor T lymphocytes recognize HLA disparity between donor and recipient tissues and respond to polymorphic and non-polymorphic host antigens, causing inflammation and damage in many organs, including the skin, liver, gastrointestinal tract, and lungs [7]. This is a medical complication characterized by a severe immune response associated with fibrosis: the formation of excess connective tissue in an organ or tissue that occurs in the final stage of inflammation [8]. Antigens are directly presented to prime allogeneic donor T-cells by antigen-presenting cells (APCs), whereas host antigens can be presented to donor T-cells by donor-derived APCs in an indirect way [9]. In GvHD pathogenesis, the conditioning regimen and tissue damage activates APCs via massive inflammatory cytokine secretion. This cytokine secretion, then, acts as an important cellular effector of GvHD progression by activating cytotoxic T lymphocytes and co-stimulatory signaling [7].
Go to :
ALLOGENEIC HEMATOPOIETIC STEM CELL TRANSPLANTATION (ALLO-HSCT)
Allo-HSCT is one of the most important organ transplant types for which GvHD has been discussed. There are many life-threatening hematologic, immunologic, and genetic diseases that affect millions of people worldwide [6]. Disruption of blood cells and hematopoietic organs is called hematologic disease and blood cell cancer, and includes various types of leukemia, including acute lymphocytic leukemia, chronic lymphocytic leukemia, acute myeloid leukemia, chronic myeloid leukemia, myeloma, lymphoma (Hodgkin's and non-Hodgkin's), genetic disorders, anemia, sickle cell disease, and conditions related to human immunodeficiency virus [10, 11]. Allo-HSCT is widely used as the only critical therapy for hematological malignant and non-malignant refractory conditions.
Allogeneic stem cell transplantation is a procedure in which healthy donor stem cells are obtained and prepared, and transferred into a patient via intravenous infusion [12]. Hematopoietic stem and progenitor cell transplantation after high-intensity chemotherapy or radiation is designed to establish hematopoiesis and immune function in patients with a variety of acquired and inherited malignancies, including leukemia, lymphoma, and myeloma, or non-malignant disorders, such as aplastic anemia, thalassemia, sickle cell anemia, and severe combined immunodeficiency [13]. During this process, a person's stem cells are replaced with new healthy stem cells that are free from contaminating tumor cells [14].
Go to :
PATHOLOGICAL PROCESS OF GvHD
The pathological process of Acute GvHD (aGvHD) in allo-HSCT is divided into four stages. In stage one, pretreatments such as high-dose chemotherapy or radiation therapy result in inflammation and the production of inflammatory cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1, -2, and -6, and chemokines. These inflammatory agents also increase the expression of adhesion molecules, co-stimulatory molecules, and MHC antigens in tissues by damaging non-hematopoietic tissues. In stage two, the innate immune cells and host APC are activated by hematopoietic graft infusion. Stage three occurs after donor-derived T lymphocytes are potently activated and proliferated by APC, thereby forming an inflammatory storm by generating and releasing a large number of inflammatory factors. Finally, in stage four, donor T-cells target host tissues, such as the skin, liver, or intestine, and initiate alloreactive cytotoxic mechanisms [6, 15].
Go to :
T LYMPHOCYTES
As T-cells are the most important factors in GvHD, current prophylaxis and treatment regimens use immunosuppressants that target T-cell subsets to mitigate the severity of symptoms. Prophylactic advances have also focused on T lymphocyte depletion, activation, and proliferation modification, key cytokine pathways, and lymphocyte traffic inhibition [6, 7].
The interaction of T cells with host antigens, and their activation following circulation in the body, causes tissue injury and is a cardinal event in aGvHD etiopathogenesis. As a result, the foundation for aGvHD prevention focuses on the depletion or modulation of donor T lymphocytes. Therefore, CNIs, such as tacrolimus and cyclosporine, plus another agent, such as methotrexate, mycophenolate mofetil, and sirolimus, that can inhibit alloreactive T-cell proliferation and activation, have been incorporated in the standard of care for aGvHD prophylaxis since the 1990s. Because of CNI-associated toxicities, including thrombotic microangiopathy (TMA) and renal dysfunction, research into prophylactic therapy with improved efficacy and less toxicity is of great importance in transplantation [7].
Regulatory T cells (Tregs), a T-cell subset that plays an important role in immunologic tolerance by releasing anti-inflammatory cytokines, such as IL-10 and transforming growth factor-β (TGF-β), can counteract the inflammatory state. Since T-helper type 2 cytokines, such as IL-4 and IL-10, can inhibit potent pro-inflammatory type 1 cytokines, such as IL-2 and INF-γ, the balance of these cytokines may govern the ultimate outcomes of inflammation; therefore, Th1 to Th2 conversion can be advantageous in aGvHD. T helper 17 (Th17) cells, another subset of CD4+ cells, can migrate to GvHD target organs and cause severe adverse effects, such as pulmonary or gastrointestinal lesions, which may play a critical role in GvHD pathogenesis.
In vivo T-cell depletion/modulation
Anti-thymocyte globulin: To reduce acute and chronic GvHD incidence by in vivo T-cell depletion (TCD), anti-thymocyte globulin (ATG), a polyclonal IgG purified from immunized horses or rabbit serum by human thymocytes or T-cell lines, has been extensively evaluated. The results demonstrated that ATG could reduce the severity of chronic graft versus host disease (cGvHD) without having a detrimental effect on overall survival rate; however, the effects of aGvHD were not reduced. Further studies have shown that excessive duration or dosing of ATG can have adverse effects, such as increasing non-rebreathing masks and relapse rates or causing immunosuppressive toxicity. Therefore, the dosing of this treatment is customized based on the total number of lymphocytes per unit of body weight, and can be used as an approach to control GvHD [7].
Post-transplant cyclophosphamide: Post-transplant cyclophosphamide (PTCy) is a game changer for GvHD prophylaxis; this procedure allows the safe and effective use of haploidentical hematopoietic stem cell transplantation (haplo-HSCT) [16]. Unlike the highly immunosuppressive regimens that can be utilized for haplo-HSCT, which lead to transplant-associated toxicity, the innovative use of PTCy treatment following non-myeloablative haplo-HSCT reduces GvHD severity [7]. T-effector cell exhaustion and Treg preservation can play an important role in the decrease in GvHD in mice [16]. PTCy can act by causing the depletion of alloreactive T-cells by eliminating their proliferation and intrathymic clonal deletion of alloreactive T-cell precursors; numerous studies have replicated these results, causing PTCy to become the most widely used haplo-HSCT regimen [17].
Sirolimus: Sirolimus is an attractive immunologic profile operator for GvHD prevention that inhibits CD8+ cells and promotes Treg proliferation in vitro. Sirolimus is a mammalian target of rapamycin (mTOR) inhibitor that synergizes with CNI to reduce Teff proliferation and activity without causing nephrotoxicity that may otherwise be caused by CNI treatment alone [18]. Although less nephrotoxic, this treatment is avoided in patients with a higher risk of veno-occlusive disease (VOD) because of its association with higher rates of VOD [19]. Additionally, in combination with CNI, sirolimus has been associated with increased rates of TMA; additionally, TMA can be resolved by discontinuation of this treatment regimen [20]. Eventually, the sirolimus/PTCy combination as a CNI-free, less nephrotoxic treatment regimen with acceptable rates of engraftment and aGvHD was assessed; consequently, it is currently reserved for conditions precluding CNI use, such as sickle cell HSCT and renal dysfunction [21].
Ex vivo TC
A clinical-grade cell separation technology has been introduced in graft manipulation for both pan and selective TCD.
Pan-TCD: Donor graft ex vivo TCD using experimental methods, such as monoclonal antibodies with or without complement, immunotoxins, or counter-flow elutriation, has been utilized to prevent GvHD due to the competing risks of relapse and non-relapse mortality. It has been has suggested that increasing rates of graft failure have been observed in TCD grafts [22, 23]. Therefore, with the availability of clinical-scale cell separation techniques, more selective methods of TCD are being explored to prevent GvHD while preserving graft-versus-leukemia (GvL) [16].
Selective TCD: For selective TCD, various solutions have been implemented; however, most of these techniques have not exhibited lasting success. Depletion of CD5+ and CD8+ T-cells was attempted for the first time in the 1990s. Although the rates of GvHD were an incentive to continue this strategy, relapse rates were high, leading to its subsequent discontinuation [16]. Using specific antibodies to deplete and modulate subset-selective T-cells has become an interesting strategy that can ameliorate GvHD without compromising the GvL effect. In contrast to α/β T-cells, γ/δ T-cells have important innate immune functions and, through α/β TCD, these γ/δ T-cells can release cytokines rapidly, thereby killing tumor and virally-infected cells without inducing GvHD [24]. Currently, the ex vivo TCD method has been established and accepted for transplant recipients. Nonetheless, all procedures, from pan to selective TCD, are associated with different limitations, advantages, and disadvantages; disadvantages include the potential for life-threatening infections but these can be blocked by the use of adoptive specific T-cells [25]. Therefore, it is hoped that, with further clinical trials, these limitations can be overcome, and this strategy can be used as an effective treatment method.
Modifying T-cell co-stimulatory/co-inhibitory pathways
Following antigen–T-cell receptor interaction and T-cell activation, we can observe the initiation of activity in co-stimulatory and co-inhibitory signaling pathways, the modulation of which can be an approach used to prevent GvHD.
Abatacept, which contains the extracellular and immunoglobulin heavy chain portions of the cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), can block CD28 and CTLA-4 as co-stimulatory and co-inhibitory T-cell receptors; this prevents CD28 and CTLA-4 from binding to the APC ligands B7-1/CD80 and B7-2/CD86 ligands, respectively, and ultimately leads to an inhibitory signal. Therefore, abatacept could be considered the most promising GvHD treatment strategy that has FDA breakthrough designation.
The OX40L blockade is another co-stimulator modifying pathway. Specifically, OX40L is a ligand for OX40 (CD134), a co-stimulatory receptor on T-cells, and a strong negative regulator of Foxp3(+) Tregs; therefore, using an antagonistic anti OX40L monoclonal antibody (moAb) may have a beneficial effect on GvHD. Consequently, this effective strategy has been gradually introduced in clinical practice [16].
Enhancing regulatory T-cells
Having a significant role in immune homeostasis, Tregs (CD4+CD25+Foxp3+) perform their suppressive function by producing TGF-β, IL-10, and IL-35. TGF-β plays a key role in inducing stem cell quiescence and promoting osteoblastogenesis, which is essential for the conservation of the HSC niche [26]. Several studies have shown that the infusion of ex vivo expanded Tregs, or their in vivo upregulation and functional enhancement after transplantation, can be beneficial for tolerance induction or GvHD prevention [16].
Extracorporeal photopheresis (ECP) as an immuno-modulatory treatment for GvHD
In ECP, a cell-based immunomodulatory treatment, the buffy coat of peripheral blood is separated and subsequently treated with a photosensitizing agent (8-methoxy psoralen). This sample, which contains leukocytes and platelets, is re-infused back into the patient after exposure to Ultraviolet A light. ECP has been shown to have various effects on different parts of the immune system. Therefore, ECP can be an effective and safe treatment for patients with GvHD without increasing the risk of relapse or infection after HSCT [27].
FTY720 improves GvHD by blocking T-cell migration and inhibiting fibrosis formation
Fibrosis, defined as excess connective tissue formation as a reparative or reactive process, is developed in the final stage of inflammatory processes like GvHD by inflammatory Th17 cells. A sphingosine-1-phosphate (S1P) analog, fingolimod (FTY720), is an effective immunosuppressive factor that accumulates T-cells in lymph nodes and blocks lymphocyte migration throughout the body via S1P1 internalization. By evaluating this treatment in an animal model, it was demonstrated that FTY720 could decrease the number of splenocytes but increase the number of T lymphocytes in mesenteric lymph nodes. Thus, FTY720 can reduce inflammation by suppressing the migration of T-cells to transplanted organs and preventing GvHD [8].
Go to :
NATURAL KILLER (NK) CELLS
NK cells are the first donor-derived lymphocyte population to recover after hematopoietic cell transplantation (HCT); specifically, this population recovery is generally observed within the first month after allo-HSCT [28-33].
The first study showed a strong relationship between GvHD progression and pre-transplant levels of NK cell activity via cytotoxic assays using herpes simplex virus type 1–infected fibroblasts as target cells; specifically, this assessment was conducted using the peripheral blood of a small and heterogeneous group of patients undergoing treatment with various protocols of HCT [28, 34].
The alloreactivity of NK cells is characterized by diverse receptors, including activating CD94/NKG2C and inhibitory CD94/NKG2A receptors, which both distinguish human leukocyte antigen E (HLA-E) [35, 36] as a non-classical MHC class I molecule [31]. According to previous studies, expression of the NKG2A receptor is continuously decreased in the early stages following HSCT; additionally, the NKG2A+ subset is reduced in patients with aGvHD after allo-HSCT [29, 31]. Therefore, NKG2A+ NK cells may play a penetrative role during the early stages of GvHD after transplantation [31].
In vitro experiments showed that NKG2A+ NK cells may hinder aGvHD by regulating T-cell immune functions. NKG2A+ NK cells inhibit T-cell proliferation and activation; moreover, T-cell apoptosis is promoted via the secretion of the inflammatory factor interferon (IFN)-γ and an increase in the number of cells with the CD4+ CD25+ FOXP3+ phenotype. However, the effect of NKG2A+ NK cells on aGvHD has not been determined [31].
In general, NK cells affect GvHD in two ways (Fig. 1). First, NK cells can suppress GvHD development via their cytotoxic function. This can be conducted directly by depleting activated alloreactive T-cells in an NKG2D-dependent manner due to the up-regulation of NKG2D ligand expression on the alloreactive T-cell population alongside inhibiting T-cell proliferation. Alternatively, this can be conducted indirectly by killing APCs, which prevents the priming of donor alloreactive T-cells; c-Kit−CD27−CD11b+ NK cells are the most potent in conducting this cytotoxic function and preventing T-cell stimulation [28, 29, 35, 37, 38]. T-cell killing by NK cells depends on both perforin production and Fas-mediated induction of apoptosis [28, 34, 39]. In an experimental study, it was demonstrated that GvHD can be induced in Severe Combined Immunodeficiency mice using IL-2-activated human NK cells that produce IFN-γ and TNF-α [28, 33, 34]. In particular, when activated, Killer Cell Inhibitory Receptor (KIR)2DS1 binds to HLA-C2, resulting in the elimination of APCs; however, KIR2DS1 can repeal the inhibition of T-cell proliferation and activation mediated by the NKG2A inhibitory receptor binding to HLA-E in humans or Qa-1b in mice. Furthermore, stimulation of NKp46 receptors on NK cells, by unknown ligands on dendritic cells (DCs), can mediate the killing of APCs during GvHD; this is because the stimulation of donor T-cells by DCs can be increased by the absence of NKp46 on donor NK cells, ultimately resulting in increased tissue damage [1].
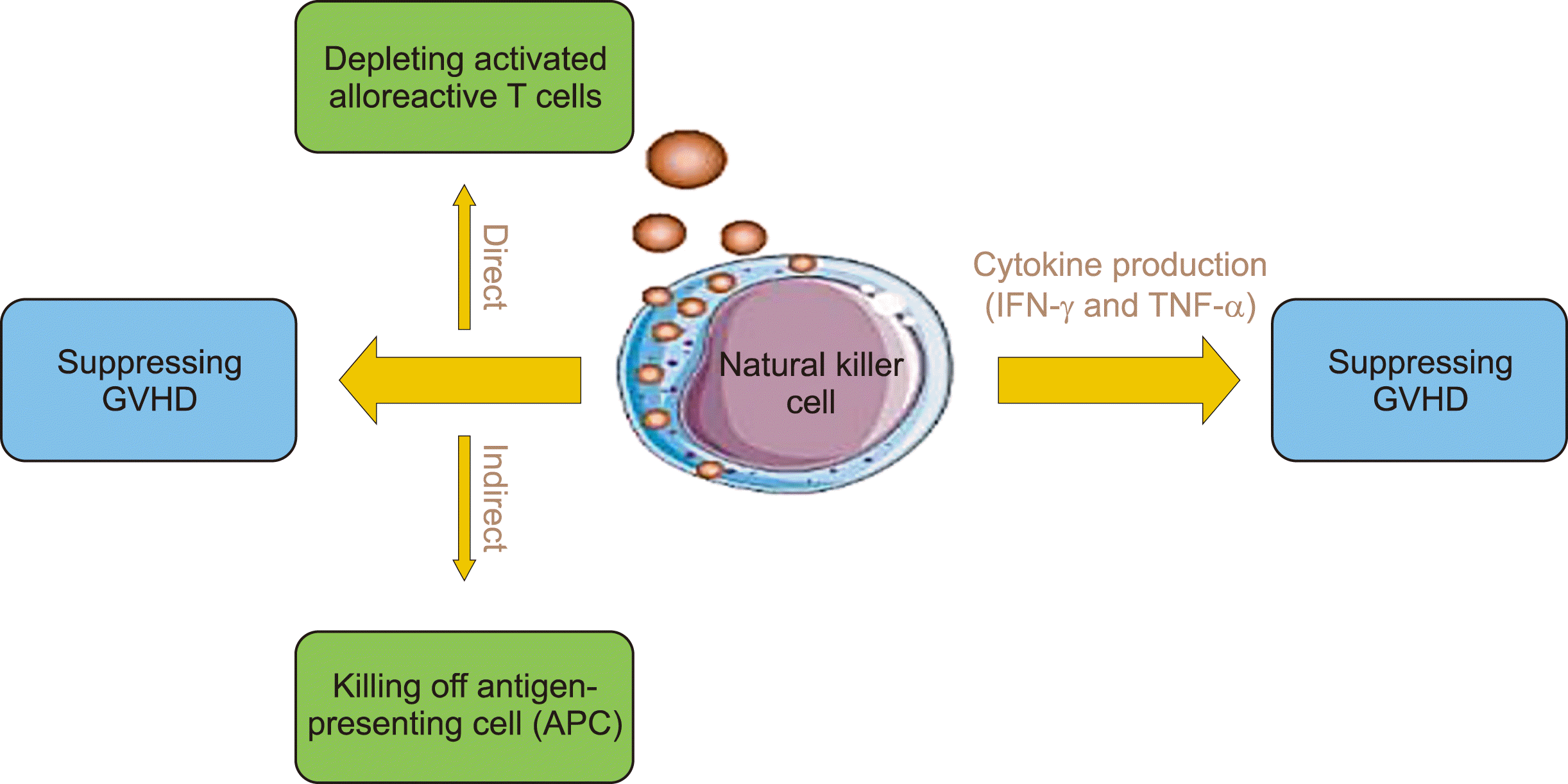
Administration of exogenous TGF-β
TGF-β is an immunosuppressive cytokine that reduces IFN-γ production and degranulation alongside decreasing the expression of activating NK cell receptors, such as NKG2D and NKp30, and decreasing total cytotoxic functions; overall, these processes can inhibit NK cells. Host Tregs are an important source of TGF-β and their depletion induces stronger NK cell–dependent allograft rejection, resulting in the suppression of NK cell activity. In contrast, exogenous administration of TGF-β prevents IFN-γ production by altering the balance between inhibitory and activating receptors and resulting in protection against GvHD [26, 39, 40]. Therefore, there is a direct relationship between increased IFN-γ levels and the incidence of GvHD [38].
Granulocyte-colony-stimulating factor (G-CSF) accelerates engraftment and increases neutrophil numbers for umbilical cord blood (UCB) transplantation, affecting group 3 innate lymphoid cells (ILC3) and NK cell differentiation [41]; additionally, before collection of HSC from donor peripheral blood, G-CSF can also be used as a potent HSC mobilizing agent [41, 42]. NKG2D expression differs among different NK cell subtypes [43]. Accordingly, several studies have indicated a postponed and reduced ILC3 expression and NK cell differentiation from HSCs that were recovered after G-CSF-induced mobilization in vitro compared to HSCs that were isolated from bone marrow (BM) or UCB. Therefore, G-CSF may affect ILC3 production [33, 41] and GvHD occurrence [33, 41, 44].
Immunosuppressive drugs
Notably, immunosuppressive drugs, such as calcineurin inhibitors, are used for the treatment of GvHD and may affect NK cell generation and differentiation. However, cyclosporine or corticosteroids may not affect helper-ILC reconstitution [41].
Conditioning regimens for stem cell transplantation
Many conditioning regimens for stem cell transplantation contain ATG, a polyclonal antibody that is derived from rabbits, horses, or pigs and targets fresh human thymocytes [45]. ATG can decrease T-cell levels following stem cell transplantation; additionally, this treatment results in functional NK cell reconstitution [45, 46] and death of targeted immune cells by induction of apoptosis, complement-mediated lysis, or NK-cell-mediated lysis [47]. As a result, adding ATG to pretreatment protocols can limit the incidence of GvHD by increasing the proportion of NK cells. Subsequently, an increase in NK cells leads to KIR-ligand mismatching, which ultimately reduces mortality rates after transplantation [45, 46, 48].
KIR2DS1 NK cells
With respect to specific genotypes, the presence of KIR haplotype B in donors has been observed to significantly decrease the incidence of GvHD [33, 49, 50]. In particular, donor NK cells expressing KIR2DS1 are effective in haploidentical allograft settings because they kill allogenic DCs and protect against GvHD [33].
Go to :
DENDRITIC CELLS
DCs originate from CD34+ HSCs in the BM and express many markers such as HLA-DR [51]. DCs link innate and adaptive immunity in response to suitable signals; consequently, this stimulation of both immunity responses can expand beneficial host responses [52].
Initiation of alloreactive T-cell responses and host DCs
DCs are APCs that recognize foreign and self-antigens and stimulate adaptive immune cells, such as effector or memory T-cells. Plasmacytoid dendritic cells (pDCs) and conventional dendritic cells (cDCs) are characterized by their remarkable capacity to generate type I IFNs upon viral infection [53]. pDCs are important effectors of the innate immune system [54]. Further, cDCs detect bacterially-derived molecules and generate pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-12p70, to stimulate T-cell subsets (Th1 and Th17) that attract and activate cytotoxic T lymphocytes [55] (Fig. 2).
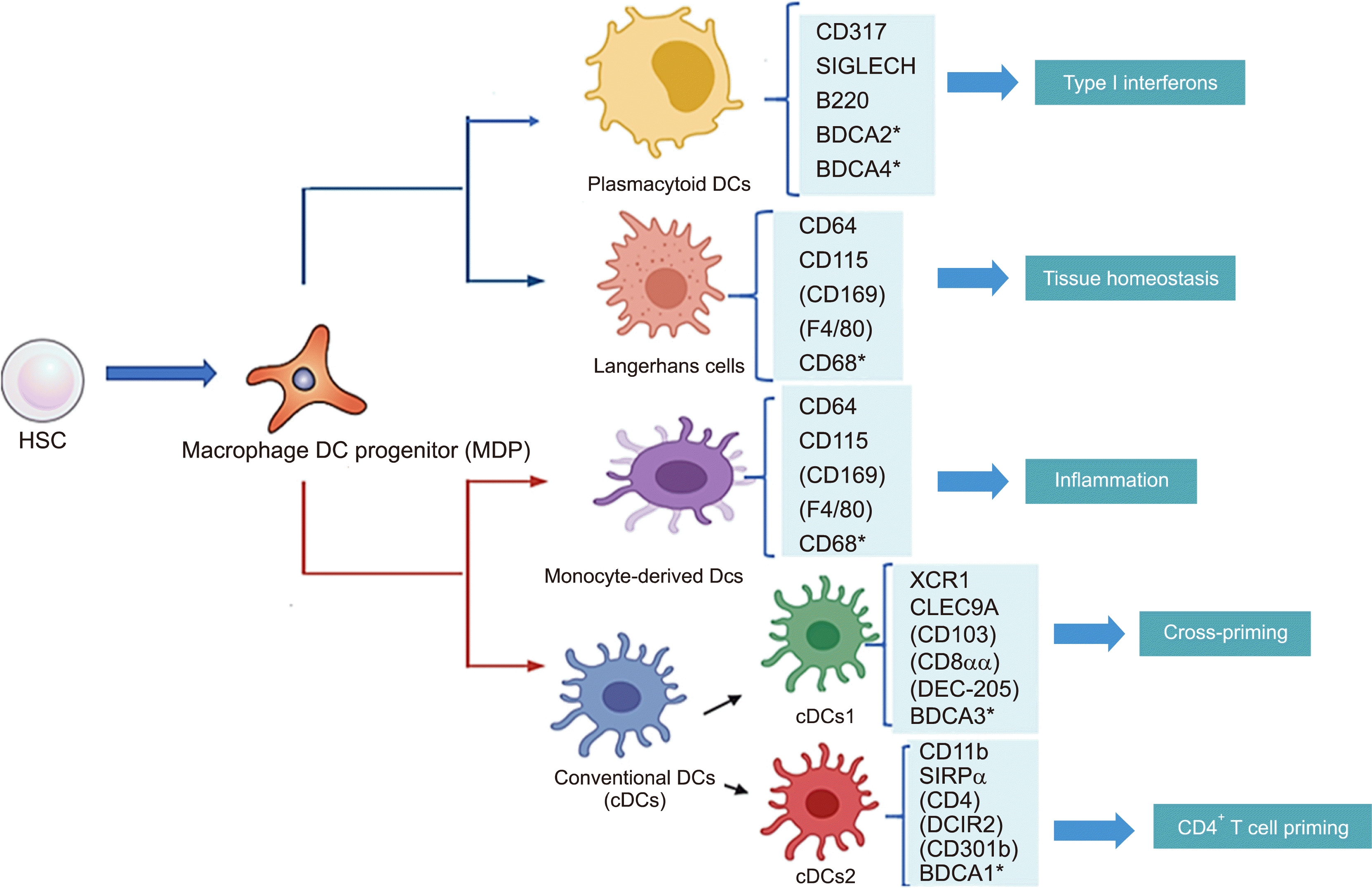
Blockade of delta-like canonical Notch ligand 4 (DLL4)
Tolerogenic, or regulatory, DCs express high levels of T-cell co-inhibitory ligands, such as programmed death ligand-1 [52]. The production of DC subsets is regulated by inflammatory stimulation. These DC-stimulating cells can selectively produce specific cytokines, such as IL-12 and IL-23, and ligands, such as delta-like 1 (DLL1) and DLL4 [55]. Immune tolerance induced by immature DCs is dependent on their low expression of CD40 and capacity to produce high levels of anti-inflammatory cytokines, such as IL-10 [56].
Production of tolerogenic dendritic cells (Tol-DCs)
Anderson et al. [57] demonstrated that host-derived DCs are important for donor T cells and are essential for the induction of disease and initiation of GvHD in MHC-mismatched transplants. DC depletion appears to be an effective way to prevent organ transplant rejection as DCs play an important role in allograft tolerance. DCs that modulate and suppress immune responses are called Tol-DCs. These Tol-DCs are characterized by a decreased expression of IL-12 and other pro-inflammatory cytokines, and an increased expression of anti-inflammatory molecules, such as IL-10 and TGF-β. Additionally, Tol-DCs can decrease T-cell proliferation and lead to T-cell apoptosis, anergy, and reduced responsiveness. This DC subset can also promote Treg induction to induce immune tolerance in transplanted cells. After allo-HSCT occurs, pattern recognition receptors induce active DCs, leading to maturation of DCs, stimulation of co-stimulatory molecules, and production of proinflammatory cytokines [58].
CTLA-4-Ig modulated DCs
Selective removal of both host- and donor-type APCs can reduce GvHD. Additionally, DCs can induce allogeneic T cell responses. Yu et al. [9] examined numerous donor DCs from the peripheral blood of 50 patients who underwent allo-HSCT. A small number of circulating DCs were found to decrease the risk of relapse and acute GvHD and could predict patient death after allo-HSCT; nonetheless, Perkey and Maillard [6] showed that DLL4 blockade can eliminate this effect, thereby preventing GvHD while preserving antitumor activity. Additionally, CTLA-4-Ig modulated DCs have exhibited positive results in suppressing immune response [56]. In vitro induction of Tol-DCs requires various stimulators and clinically approved drugs, including rapamycin, dexamethasone, vitamin D3, IL-10, and TGF-β [58]. Stenger et al. [59] evaluated a polyclonal ATG antibody with various immunologic effects, including several impacts on DCs, such as inhibiting antigen uptake and maturation, decreasing antigen-presenting capacity, and inducing complement-mediated lysis of these cells.
Histone deacetylase inhibitors
Histone deacetylase inhibitors reduce DC numbers through Toll-like receptor–induced co-stimulatory molecule presentation, pro-inflammatory cytokine release, and T-cell allo-stimulatory activity. There are numerous open clinical trials surrounding the efficacy of cellular therapies and biological interventions for the treatment or prevention of GvHD [59].
Genetically modified DC
Immature DCs have been genetically modified to express soluble TNF-α receptor I and C-C chemokine receptor type 7 on GvHD and GVL in allogeneic BM transplantation mice. Studies have shown that pDCs inhibit proliferation and activate autologous T-cells via a type I IFN signaling–dependent mechanism. Alternatively, GvHD prevents the regeneration of tolerogenic donor pDCs by depleting DC progenitors, rather than inhibiting pDC maturation. The uncontrolled production of functional donor DCs is a challenge for GvHD [60]. DCs can be targeted to reduce the effects of GvHD and the reactive T-cell response. For example, the co-transfer of tolerogenic DCs and Tregs may be more useful in reducing GvHD reactions in vivo. However, one major challenge for this treatment is the need to produce a large number of donor-type tolerogenic DCs with sufficient time to exert their function [9]. Tol-DCs are a small group of DCs that are distinguished by low expression levels of co-stimulatory agents and pro-inflammatory factors. Tol-DCs stimulate immune tolerance by inducing Treg proliferation and inhibiting T-cell activation, resulting in a reduced effect of GvHD.
Go to :
MESENCHYMAL STROMAL CELLS
Mesenchymal stromal cells (MSCs) are multipotent precursor cells with heterogeneous cell populations in all organs of the body and were first found in the BM in 1968. These cells are derived from an array of embryonic and adult tissues in the human body [60].
MSCs secrete a variety of cytokines and regulatory molecules that decrease host defenses by modulating immune effector cells and proinflammatory immune responses. These MSCs also secrete a large number of soluble factors; these factors are known as the “secretome” and include indoleamine 2,3-dioxygenase, that plays an essential role in depleting tryptophan, alongside kynurenine, prostaglandin 2, IL-10, TGF-β, nitric oxide, HLA-G5, the highly anti-inflammatory molecule TNF-α-induced gene/protein 6, hepatocyte growth factor, vascular endothelial growth factor, bFGF, insulin-like growth factor 1, chemokine (C-C motif) ligand 2, epidermal growth factor, platelet-derived growth factor, IL-6, and SDF-1 [25]. Additionally, MSCs induce perforin-dependent apoptosis in recipient cytotoxic T cells (Fig. 3) [60].
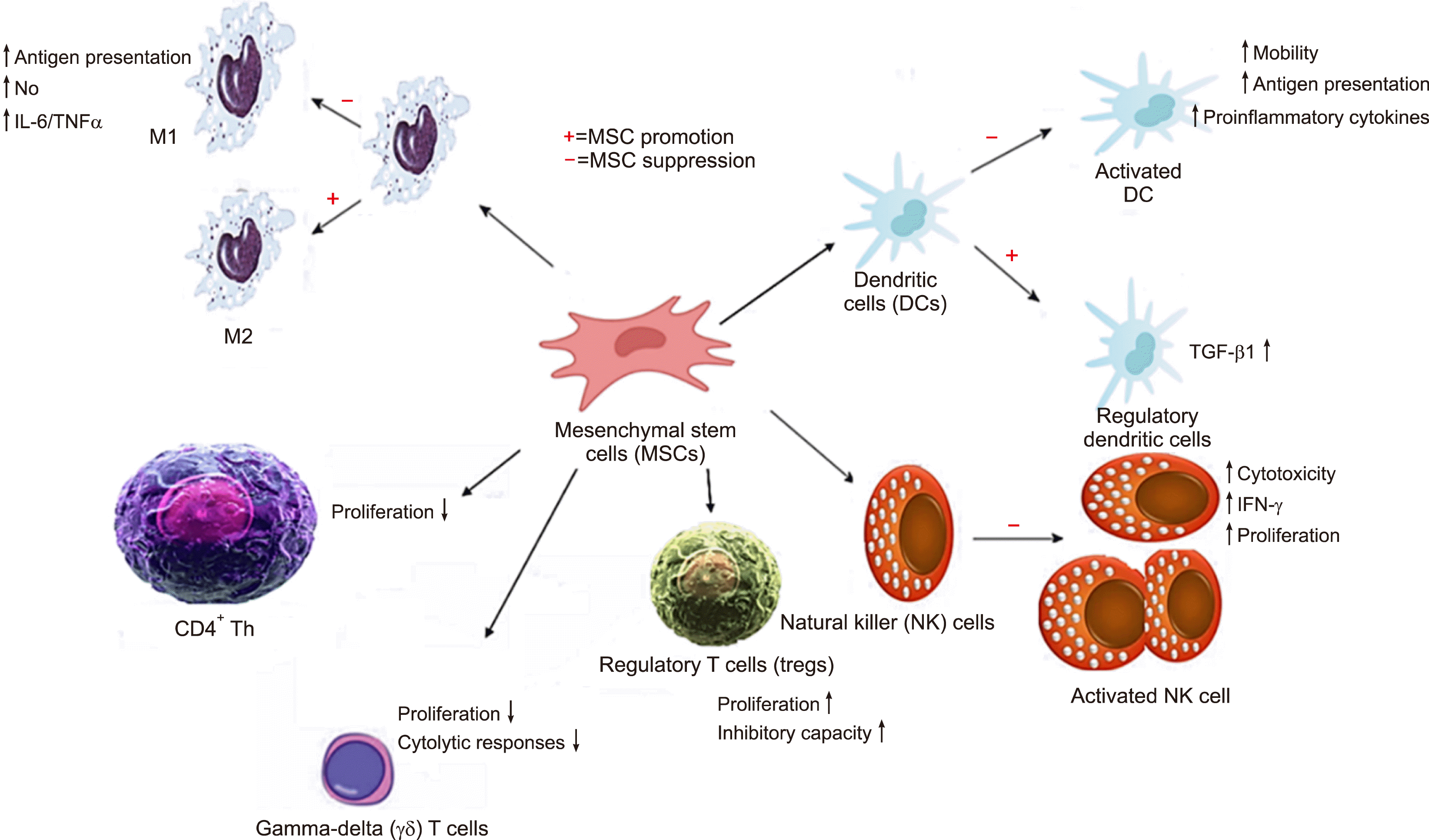
MSCs are highly immunosuppressive upon stimulation with inflammatory cytokines. However, MSCs can also enhance immune responses. Some studies have shown that MSCs cannot protect mice from GvHD, although treatment with MSCs did suppress lymphocyte proliferation in vitro to some degree [61].
Ringdén et al. [62] demonstrated that co-transplantation of HCT and MSCs prevented the development of lethal GvHD in the treatment of six children with decidual stromal cells (DSCs). Further, MSCs derived from different tissues are well-tolerated and have shown promising outcomes for acute GvHD in patients.
MSC-derived exosomes
MSC-derived exosomes have streamlined regulatory approval and commercialized products for the treatment of GvHD, alongside a diverse range of inflammatory conditions [63].
MSCs can facilitate HSC engraftment via paracrine mechanisms, including secreting mediators and hematopoietic cytokines and suppressing the remaining host immunity [64]. The first patient with steroid-refractory aGvHD was treated with allogeneic MSCs in 2004 [65]. Giebel et al. [54] showed that MSC-derived extracellular vesicles (EVs) indirectly block the inhibitory effects of T lymphocytes and control the improvement of GvHD symptoms. Nonetheless, it is unclear whether the anti-inflammatory abilities of EVs are associated with high levels of TGF-β, IL-10, and HLA-G.
EV therapy of UCB stem cells has been observed to induce an increase in expression of C–X–C chemokine receptor type 4, a key part of HSC niche, and induce the completion of CD34+ cell migration from the peripheral blood to BM niche. Winer et al. [66] indicated that BM-MSC-EVs alleviated symptoms in a treatment-resistant, grade IV aGvHD patient. Consequently, many researchers have designed strategies to modify and improve MSC therapy in GvHD [64].
MSCs transduced with IL-10 or through exposure to IFN-γ and nitric oxide
Other studies in mice have shown that MSCs inhibit GvHD via IL-10, or IFN-γ with nitric oxide [40]. MSCs, which can be extracted using various methods, have features that are not found in other cells. Several years of research has demonstrated that MSCs are important cells that interact with their immediate surroundings and adjacent cells, thereby supplying cell-based reactions that can be utilized in therapeutics [67]. Several studies have been conducted on the co-transplantation and co-culture of MSCs with HSCs to improve HSC expansion and differentiation. These studies evaluated the clinical capacity, inhibition, and potential treatment of GvHD in HSCT; however, the corresponding results of these studies revealed different effects in this treatment. In addition, there are several disadvantages to the clinical usage of MSCs, such as pneumonia-related death in allo-HSCT patients, increased tumor expansion, pulmonary embolism due to unregulated differentiation of MSCs, and ectopic tissue formation [68]. Nonetheless, MSCs are observed to be immunosuppressive in vitro, in preclinical animal models, and in clinical studies. An advantage of MSC use in GvHD treatment is their high safety with few side effects [68]. Ringdén et al. [62] found that DSCs have immunosuppressive activity and in vitro alloreactivity when used in combination with lymphocyte culture. DSCs are also more effective than BM-MSCs in treating acute GvHD; however, an advantage of MSCs, compared to DSCs, is their toxicity profile. Some recent studies have used MSC-EVs, because of their additional benefits, for cellular therapy in allo-HSC grafts, engraftment, and inhibition of GvHD. Application of MSC-EVs improved the effects of HSCs, including enhancing self-renewal, inhibiting differentiation, promoting homing for HSC expansion, and preventing GvHD following HSCT [62].
Go to :
CONCLUSION
To establish therapeutic solutions for GvHD, it is necessary to understand the cells involved in this process and the immunological basis of the corresponding mechanism.
In this review, we identified strategies that have the potential to become preventive treatments for GvHD pathogenesis. Among the aforementioned cases, different solutions have been investigated to reduce the effect and function of T lymphocytes, which play a central role in GvHD progression.
In these therapeutic strategies, attempts have been made to control the activity and proliferation of T-cells by altering cytokine levels or signaling pathways. In this field of treatment, several promising strategies have been discussed: modulation of co-stimulatory pathways, cytokine targeting, targeting lymphocyte trafficking, inhibition of key canonical pathways, and TCD.
Although many studies have been conducted to determine the role of NK cells in GvHD, the role of these cells remains unclear. NK cells play both negative and positive roles in GvHD due to subset variations at each stage after HSCT and their heterogeneity; this understanding explains the emergence of contradictory evidence across different experimental approaches. Nevertheless, clarifying the precise effect of NK cells on GvHD induction requires further elucidation, thereby allowing optimization of cell production procedures to improve allogeneic NK cell antitumor potential and prevent GvHD induction. In contrast, DCs have shown great potential for the treatment of autoimmune diseases and tissue transplantation [69].
Alternatively, several studies have demonstrated that, because of their immunosuppressive properties, MSCs play an essential role in preserving the regulation of immune tolerance, preventing organ transplantation, autoimmune treatment, and tumor escape. Additionally, MSCs are also useful in reducing GvHD symptoms and improving graft survival [70].
MSC treatment increases Treg cell numbers in the spleen of graft recipients; consequently, this results in decreased infiltration of healthy tissues by donor T-cells, enhanced CTLA-4 expression, and reduced CD80/86 expression on DCs, ultimately suppressing the incidence of GvHD [71].
Considering that traditional methods are still used to control the pathogenesis of GvHD, it is anticipated that, with further research, new techniques will be established that can minimize the complications of GvHD and retain the benefits of GvL (Fig. 4).
Go to :
 XML Download
XML Download