

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Acute lymphoblastic leukemia (ALL) is a type of blood cancer that is the most frequent cancer in infants [1, 2]. In ALL, different chromosomal alterations have been identified that are associated with clinical characteristics and response to treatment [2]. However, the overall survival is 25–30% in adults with ALL, unlike pediatric patients where survival is much higher [3, 4]. The use of cell therapy based on CAR cells in developing countries is expensive and unlikely [5]. Searching for alternative therapeutic strategies for this disease is one of the objectives to improve the quality of life of patients. Induction of mitotic death in cancer is one of the strategies explored. Mitotic death is defined as a specific variant of regulated cell death caused by mitotic catastrophe [6, 7]. Mitotic catastrophe is a biological process that prevents cell survival. Different pathways that activate mitotic catastrophe have been described, one of which is through p53 and another through the RAS pathway [8-10].
RAS proteins belong to a family of GTPases that activate several cell signaling pathways, such as proliferation, differentiation, and survival [11]. The high prevalence of Ras mutations in human cancer has been recognized for many years [12]. However, there is also evidence that supports a paradoxical role of Ras in the suppression of tumorigenesis and initiation of cell death [13]. The induction of different mechanisms of cell death, such as apoptosis, mitotic catastrophe, autophagy, and methuosis, has been observed after ectopic expression of activated Ras [14, 15].
The Ras protein is a central component of mitogenic signal transduction pathways and is essential for both the quiescent state exit and the G1/S transition of the cell cycle [16, 17]. The loss of viability has been associated with sustained mitogenic signals and aberrant progression of the cell cycle, leading to replicative stress, increasing the possibility of generating alterations such as gene amplification and aberrant chromosomes within a simple cell cycle [18].
In studies on rat fibroblasts [19] or normal thyroid cells [20], different effects of activated Ras have been observed on the cell cycle, with arrest in the G1 or G2/M phases preceding apoptosis. Ectopic expression of Ras and ARH1, a member of the Ras family, is involved in cell cycle arrest in S/G2/M and S phases, respectively [21-23].
This study aimed to investigate the effects of H-Ras gene transfection on the cell cycle and morphology of MOLT-4 acute lymphoblastic leukemia cells.
MATERIALS AND METHODS
Cell culture and transfection
MOLT-4 cells CRL-1582 (a kind gift from Dr. Carl Miller, UCLA School of Medicine, USA) were grown at a density of 5×105 cells per dish in 5 mL of RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 incubator. These cells were authenticated by the ATCC Cell Authentication Human STR Testing Service (ATCC, Manassas, VA, USA), and all experiments were performed with mycoplasma-free cells. The cells in the exponential phase were electroporated with linearized DNA of the Raswt/neo plasmid, Rasmut/neo, or pSV2-neo. At 24 h after transfection, the cells were purified using Ficoll-Hypaque. After 24 h, G-418 was added at 600 µg/mL.
Cell morphology and cell cycle analysis
The cells were spread on slides 96 h after transfection and stained with Wright. Cell viability was evaluated at different time points using the trypan blue exclusion assay. For cell cycle analysis, transfected cells were fixed for 96 h with 80% cold ethanol, treated with RNAse A (100 µg/mL), and stained with propidium iodide (50 µg/mL). The DNA content of cells was analyzed using a FACS Calibur flow cytometer (Becton Dickinson Immunocytometry Systems, Franklin Lakes, NJ, USA). Data analysis was performed using Cell Quest (BDIS) and ModFit software (Verity Software House Topsham, ME, USA).
Western blot assay
Cells were lysed using 150 mM NaCl, 50 mM Tris (pH 7.5), 0.5% Nonidet P-40, and complete protease inhibitor (Roche Applied Science). The proteins were separated in 12.5% polyacrylamide gels (30:1) (acrylamide:bisacrylamide) and electrophoretically transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat milk in PBS-Tween 20 and incubated with goat anti-actin polyclonal antibody, anti-PCNA mouse monoclonal antibody, and anti-cyclin D1 (1:500) rabbit polyclonal antibody (Santa Cruz, CA, USA), followed by goat anti-mouse or anti-rabbit antibodies conjugated to horseradish peroxide.
Cytogenetic analysis
Colcemid (0.5 mL) was added to the cells 48 h after transfection. The cells were incubated for 4 h at 37°C and then placed in hypotonic solution (0.075 M KCl) for 20 min. Then, the cells were fixed with methanol-acetic acid (3:1) and placed on the slides by dripping. The chromosome number was determined using 100 metaphase analyses.
Real-time polymerase chain reaction (PCR)
H-Ras expression was detected using real-time PCR. The expression levels of the H-ras (Hs00978050) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Hs00985689) genes were measured using TaqMan gene expression assays (Step One of Applied Biosystems. Foster City, CA, USA). The mRNA level was normalized to GAPDH expression in each sample and presented as the ΔCt value (ΔCt=Ct GAPDH-Ct target mRNA). A lower ΔCt value indicated lower expression of the target mRNA.
Statistical analysis
Comparisons between the means of the cell cycle data were made using Student’s t-test for paired data. Data on cell death, chromosome number, and H-Ras expression were analyzed using the ANOVA test. Additionally, in the case of chromosome number data analysis and H-Ras expression, the Scheffe test for multiple comparisons was used.
RESULTS
H-Ras alters nuclear stability
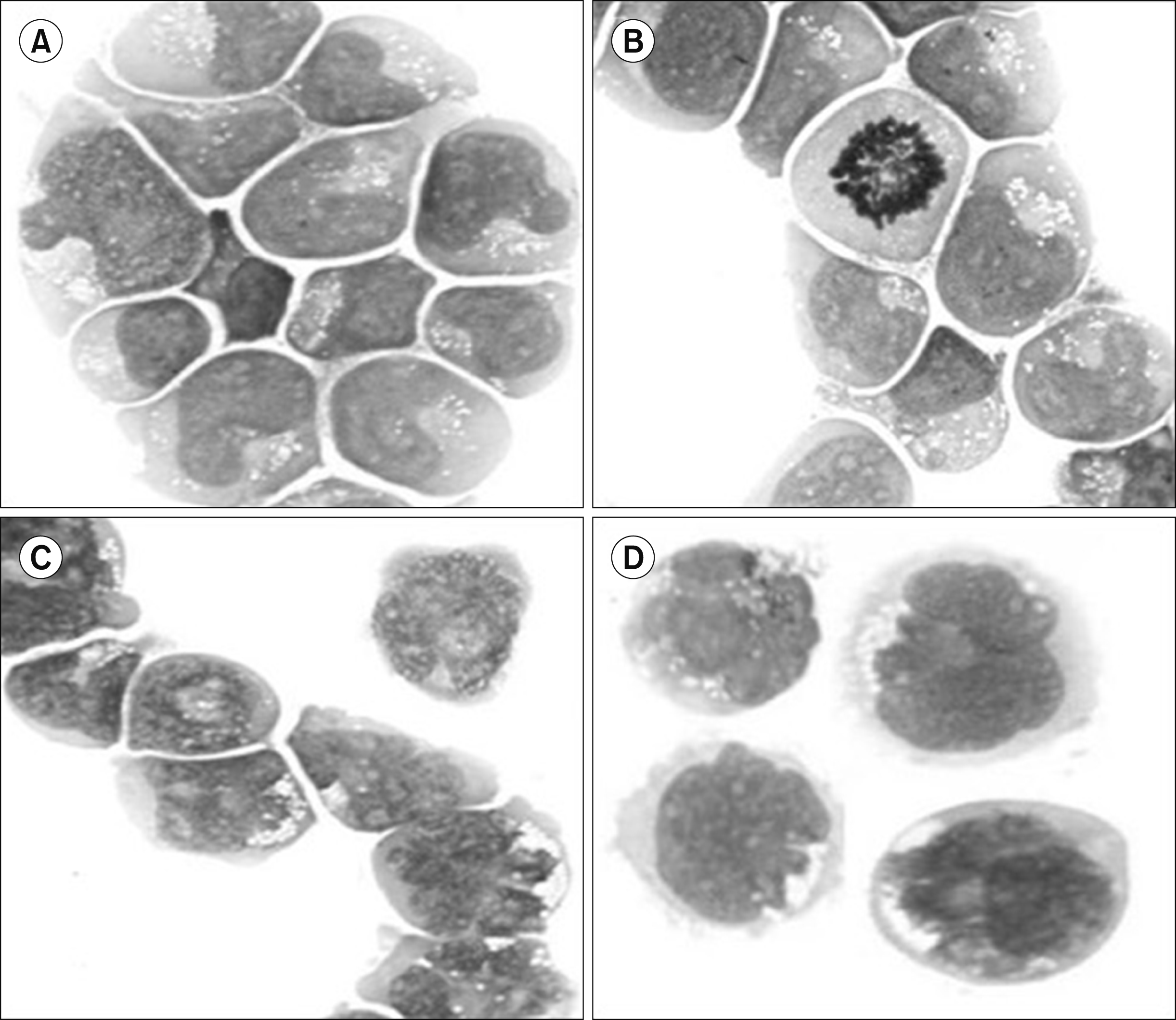
MOLT-4 cells were transfected with either normal or mutated H-Ras genes, linked to a neoresistance gene. Selection with G-418 (antibiotic ensures effective positive selection for cells expressing the neomycin resistance gene). Cell morphology was analyzed. Ras-transfected cells showed multiple nuclei (Fig. 1C, D) and micronuclei.
H-Ras induces cell cycle S-phase arrest
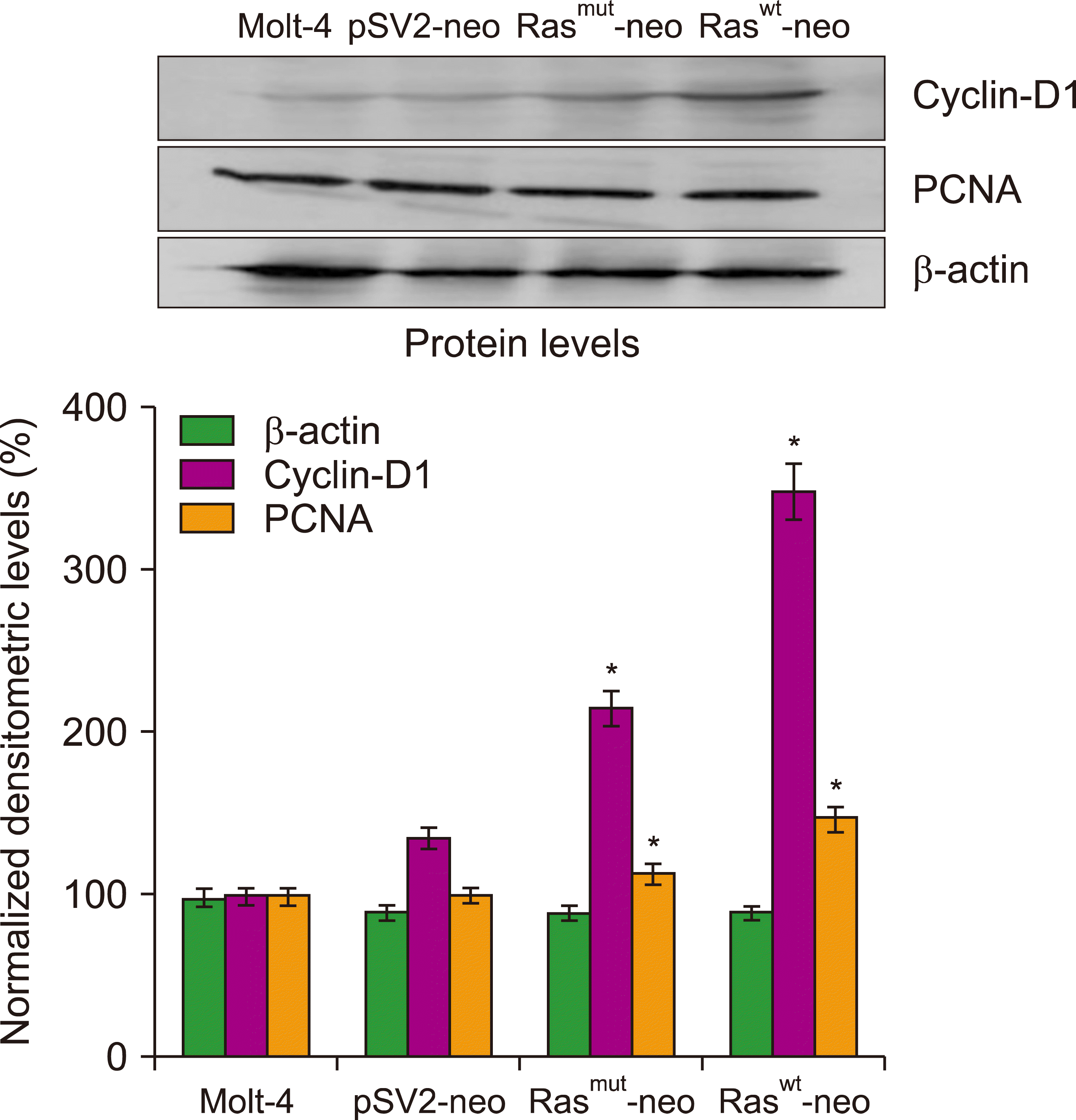
The distribution of cells in different phases of the cell cycle was also analyzed. The cells were transfected and analyzed by flow cytometry at 96 h. Transfection of MOLT-4 cells with normal or mutated H-Ras resulted in cell cycle S-phase arrest (Fig. 2). The percentages of cells transfected with either the normal or mutated H-Ras gene in the S-phase was 83.6% (P<0.0005) and 81.7% (P<0.0001), respectively, compared with untreated cells or those that were transfected with the vector, which had percentages of 45.6% and 31.3%, respectively. These results indicate that the transfection of the H-Ras gene induces cell cycle S-phase arrest and that these cells cannot proceed to the G2/M phase. Overexpression of cyclin D1 was induced by activated Ras [24] and this protein can prolong the S-phase and inhibit growth. The expression of cyclin D1 and PCNA proteins was analyzed in H-Ras transfected cells. Increased cyclin D1 levels were observed in cells transfected with either the normal or mutated gene at 72 h (Fig. 3). These results indicate that Ras transfection induces overexpression of cyclin D1 and is a possible mechanism that causes cell cycle S-phase arrest.
Increased number of chromosomes in Ras-transfected cells
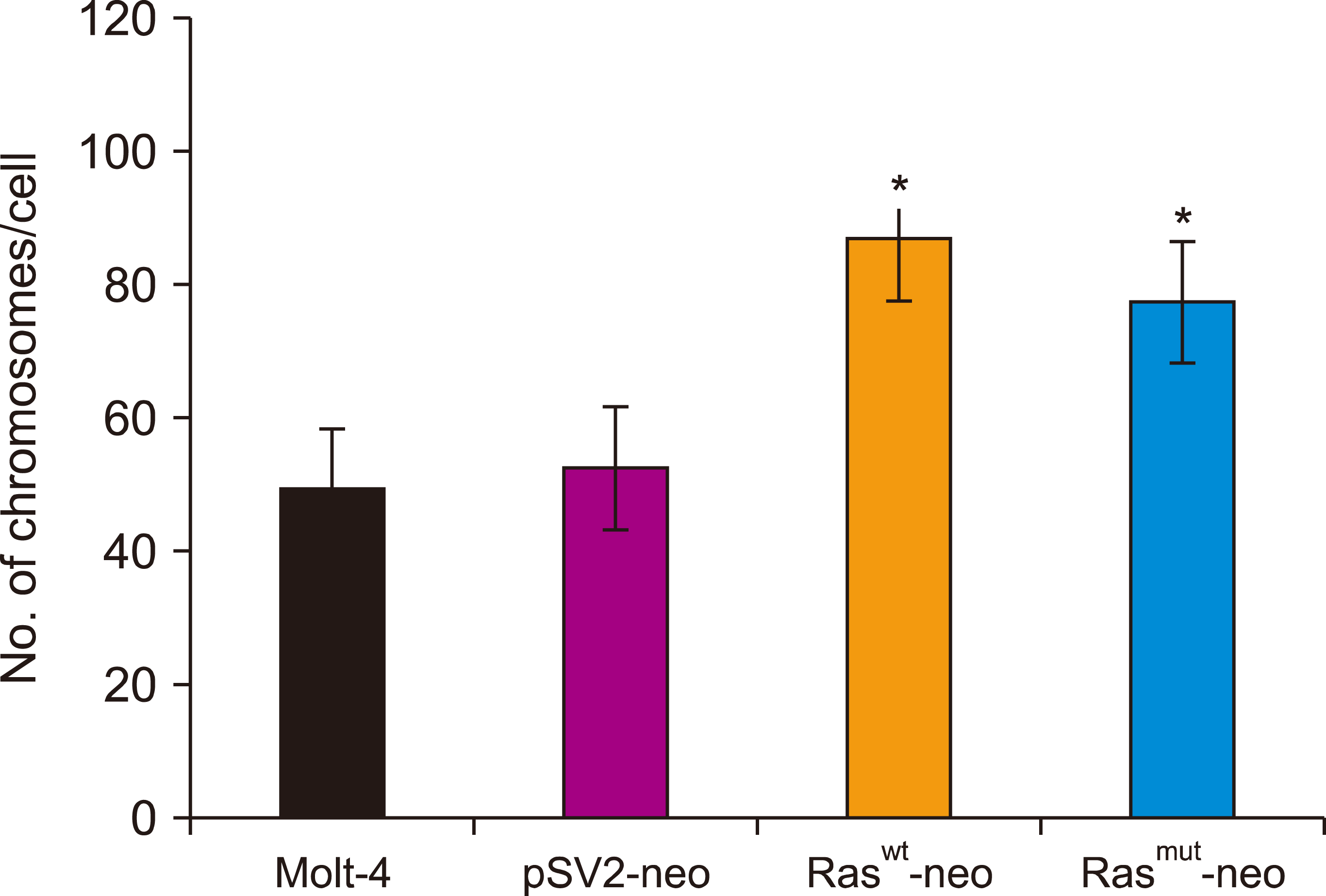
The MOLT-4 cells were heteroploid with an average of 49 chromosomes per cell, a range of 12–136, and a mode of 22 (Fig. 4). Cells transfected with the vector alone (pSV2neo) showed no significant differences (average=52; range=10–130; mode=35). However, cells transfected with normal or mutated H-Ras genes exhibited a significant increase in chromosome number: average=86, range=40–151, mode=92; and average=77, range=31–180, mode=76, when compared with the vector alone and Molt-4 cells without transfection, respectively (P<0.001). These results indicate that transfection with Ras affects the integrity of the mechanisms that maintain the correct number of chromosomes per cell.
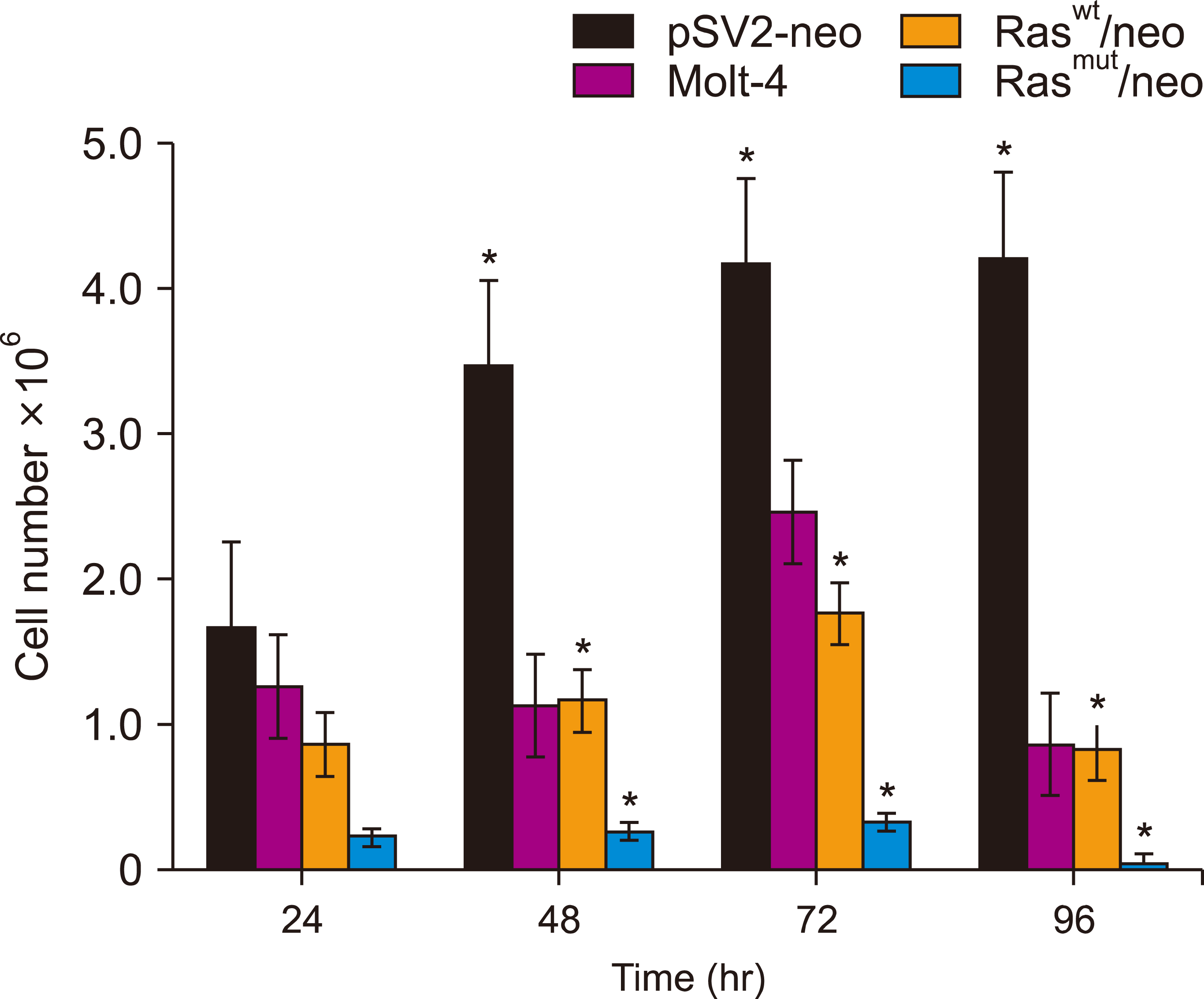
H-Ras induces cell death
The Ras-induced effects on cell viability were analyzed (Fig. 5). The cells were transfected with H-Ras, and their viability was analyzed at the indicated times. The number of cells treated with the mutated H-Ras gene at 96 h was extremely low compared with that in cells transfected with the neo gene (P<0.05) and corresponded to cells that were found mainly in the S-phase of the cell cycle. Conversely, the normal H-Ras gene-transfected cells were arrested in the S phase, similar to those transfected with the mutated gene (Fig. 2). However, the viability of normal H-Ras gene-transfected cells was less evident than that of cells transfected with the vector alone (P<0.05) (Fig. 5).
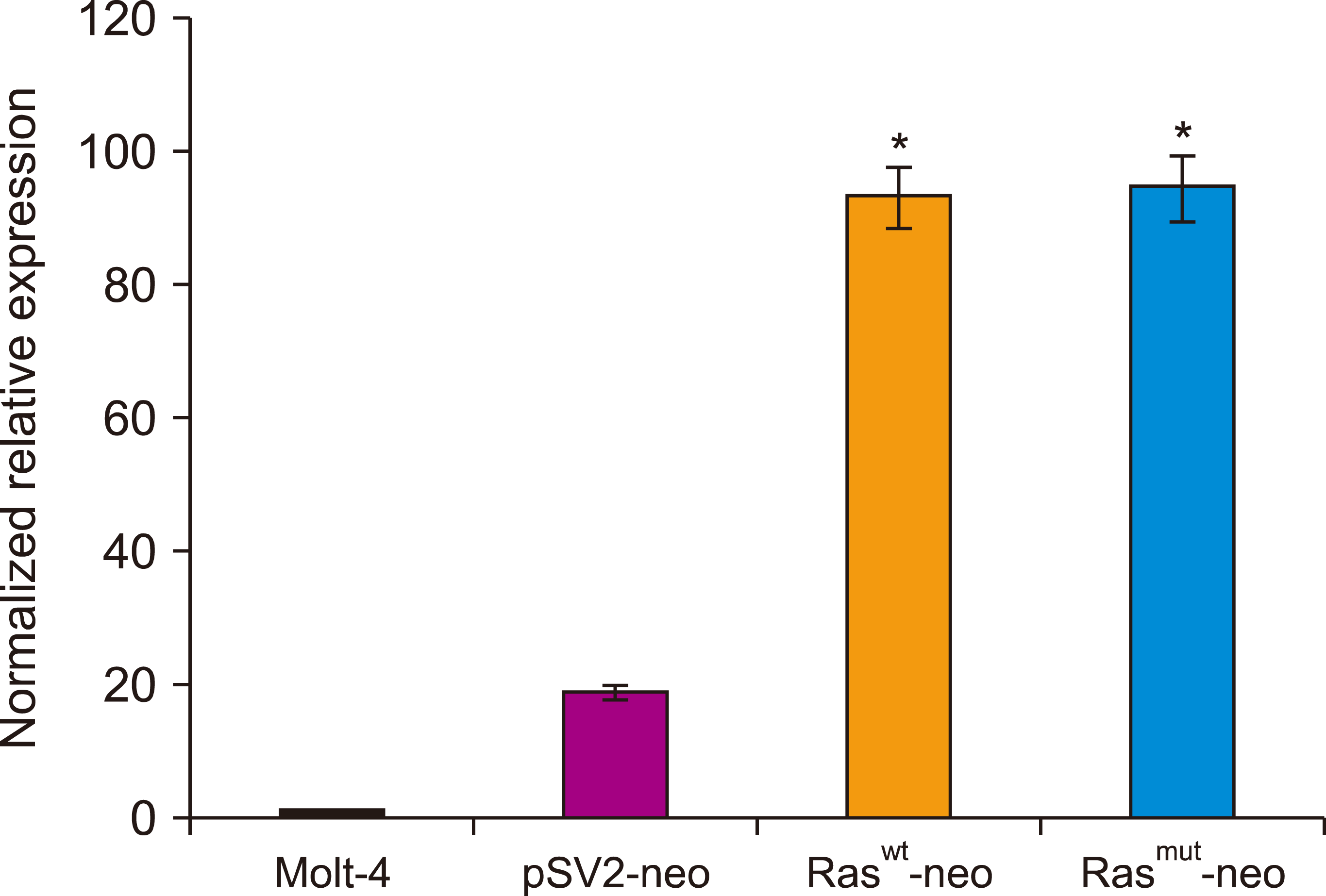
Increased H-Ras expression in transfected cells
To ascertain whether the effects observed after transfecting MOLT-4 cells with Ras were due to abnormal gene expression, real-time RT-PCR was performed. Increased H-Ras expression was detected in transfected cells (either normal or mutated genes) (Fig. 6), compared with untreated cells or cells transfected with vector alone (P<0.05). Perhaps, this is the mechanism responsible for the observed alterations induced by transfection of the Ras gene.
DISCUSSION
In this study, we analyzed the effect of transfection of the Ras gene, either normal or mutated, on MOLT-4 cells. We found that overexpression of Ras-induced alterations in cell morphology, cell cycle, and number of chromosomes eventually culminated in mitotic catastrophe of cells.
Mitotic catastrophe is a mechanism that prevents the proliferation of cells that are unable to complete mitosis due to alterations and failures in the control of the cell cycle. Morphologically, cells show nuclear changes, including multinucleation, macronucleation, and micronucleation. Although the molecular mechanisms are not well known, it has been noted that they can be caused by different factors, including exogenous factors such as xenobiotics that alter replication, cell cycle control points, chromosomal segregation or microtubule dynamics, and endogenous factors, among which are elevated levels of replicative stress or mitotic stress caused by aberrant ploidy or deregulation in the expression or activity of replication factors. Primary alterations that induce catastrophic mitosis can originate in other phases of the cell cycle, including the S phase [18, 25].
As previously reported for HeLa cells, we observed that, after transfection with Ras, the cells presented multiple nuclear alterations (multi-, macro-, and micronucleation) to finally die.
Nuclear alterations and cell cycle arrest promoted by mitotic stress induced by the activation of mutated H-Ras have been reported in primary human fibroblasts [18]. This process culminated in extended mitotic arrest and aberrant exit via mitotic slippage, generating multinucleated senescent cells.
Mitotic catastrophe is one of the main mechanisms of cancer chemotherapy. In NK cell lymphomas, bortezomib, at a concentration used in myeloma cells, induces apoptosis; however, in some cell types, a higher concentration of the drug was required, leading to death of these cells through a mitotic catastrophe [26]. Cell cycle arrest in the G2/M phase and subsequent mitotic catastrophe have been reported in lymphoma cells that have greater sensitivity to bortezomib [27] or are resistant to rituximab [24]. Damage to DNA with cisplatin induces two different modes of cell death in ovarian carcinoma cells: when the cells + p53, + Chk2, and caspase-2 are present, the cells are directed toward apoptosis; in the absence of functional p53 and lack of Chk-2, damaged cells undergo mitotic catastrophe [28]. Cell cycle arrest in the S/G2/M phase and induction of mitotic catastrophe in lung tumor cells have also been reported after treatment with ophiopogonin B [29] and in HeLa cells after the treatment with methyl-amino levulinate, a photosensitizer used in photodynamic therapy [30]. We observed that MOLT-4 cells transfected with either normal or mutated Ras were arrested in the S phase of the cell cycle. It would be helpful to determine whether the same phenomenon occurs in other cell types. For this reason, the cell lines derived from T-acute lymphoblastic leukemia, such as JURKAT and DND-41, will be transfected with either normal or mutated H-Ras genes and then analyzed by FACS. These results may contribute to the development of novel therapeutic strategies.
Conversely, it has long been known that Ras activation induces cyclin D1 overexpression [31, 32], which in turn can inhibit DNA synthesis by binding with a critical regulator of the S phase of the cell cycle, PCNA [33].
Cyclin D1 overexpression has been directly linked to an increase in the length of the S phase [34] and the entire cell cycle [35]. A delay in progression through the S phase has been reported in cells treated with zidovudine, which could be due to a longer time for repair and termination of the S phase [36]. In fact, it has been described that mitoxantrone induces cell cycle arrest of MOLT-4 cells in the S/G2/M phase [37, 38]. Treatment of human TK6 lymphoblast cells with hydroquinone causes an arrest in the G1 phase of the cell cycle, which transits into an arrest in the S phase as H-Ras expression increases [39]. Ras induces replicative stress and DNA damage in fly cells and human fibroblasts [40]. Overexpression of mutated Ras causes an increase in TBP expression, which induces an increase in RNA synthesis. The abovementioned mechanism, together with the accumulation of R-loop, results in a delay of replication forks and DNA damage [41]. Moreover, mutated Ras promotes hyperreplication through the overuse and reuse of replication origins during the S phase of the cell cycle, causing replicative stress and genomic instability [42].
Furthermore, H-Ras wild-type gene can suppress the early stages of tumorigenesis in mouse pancreatic cancer cells infected with modified retroviruses containing the gene [13]. In the present study, transfection and subsequent overexpression of H-Ras induced aberrant cell cycle progression and mitotic catastrophe. We believe that the death of MOLT-4 cells after H-Ras transfection was caused by mitotic catastrophe and apoptosis. As part of our research, we measured apoptosis by Western blot analysis to explore the levels of caspases 3 and 9 in MOLT-4 cells and other T-acute lymphoblastic leukemia cell lines (JURKAT and DND-41) transfected with either normal or mutated H-Ras genes to reinforce our findings [43].
Introduction of mutated H-Ras into normal human fibroblasts induces arrest in the G1 phase of the cell cycle and senescence if p53 is normal [44]. Mouse embryonic fibroblasts lacking p53 are arrested in the S/G2 phase and die by mitotic catastrophe after irradiation. However, cells that contain normal p53 are also arrested but have a low frequency of mitotic catastrophe [45]. HeLa cells have been shown to harbor transcriptionally active sequences from human papillomavirus type 18, including the E6 oncogene, which mediates p53 degradation [46]. The following mutations in MOLT-4 cells have been described: p53, PTEN, and STK11 [47]. All of which are tumor suppressor genes that would normally arrest the cell cycle and activate apoptosis [48, 49]. However, in these cells, the sustained activation of MAPKs after transfection of H-Ras cannot lead to apoptosis due to the aforementioned mutations; thus, the cells died through a mitotic catastrophe.
MOLT-4 cells were established from a patient with T-ALL who was treated with vincristine, 6 mercaptopurine, and prednisone. However, we believe that mutations in p53, PTEN, and STK11/LKB1 prevented cell cycle arrest and subsequent cell death via apoptosis. There was no response to treatment and the patient relapsed. The cell line was established before the patient’s death. In the present study, these cells were transfected with Ras, promoting death by mitotic catastrophe.
It is of great interest to try to eliminate tumor cells by using substances that target proteins that control the cell cycle [50]. Cancer cells show an increase in replicative stress due to the activation of oncogenes, which provides opportunities to induce death by inactivating residual compensatory mechanisms. Therefore, we suggest that in patients who present resistance and relapse to antitumor treatment, mitotic catastrophe should be induced in tumor cells. The most frequent examples of tumor promoter aberrations that increase replicative stress include mutations that affect the Ras signaling pathway, Rb pathway disorder, amplification and overexpression of c-Myc or cyclin D1 and E, and haploinsufficiency of p27Kip1. The search for drugs that can induce the death of tumor cells with such vulnerabilities is currently under investigation. One possibility is through the activation or repression of target genes, such as Ras. For example, overexpression of aquaporin-9 (AQP9) induces the activation of Ras and the subsequent arrest in the S-phase of the cell cycle, increasing the effectiveness of chemotherapy during colorectal cancer treatment [51].
CONCLUSION
We analyzed the effect of abnormal expression of Ras. Transfection of MOLT-4 cells with Ras genes induces alterations in cell morphology, including arrest in the S phase of the cell cycle and changes in the number of chromosomes, which eventually culminate in the mitotic catastrophe of cells.
 XML Download
XML Download