

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Sarcopenia is defined as a progressive and generalized skeletal muscle disorder involving the accelerated loss of muscle mass and function. Sarcopenia can result from multiple pathologies; these include aging-associated reductions in muscle mass, cachexia caused by malignant tumors, muscle wasting resulting from acute diseases, and a decrease in muscle mass due to hereditary myopathy [1]. Sarcopenia is associated with adverse outcomes including falls, functional decline, frailty, reduced mobility, diminished quality of life, and fall-related injuries such as fractures, which can require costly hospitalization and extended rehabilitation, and may increase mortality.
The most widely cited definition of sarcopenia is from the European Working Group on Sarcopenia in Older People; based on this definition, large-scale studies suggest the prevalence of sarcopenia is 6% to 15% of the general population. Emerging evidence demonstrates that the incidence of sarcopenia increases with age: for example, one study showed an incidence of 1.6% in people aged 40 to 79 years and 3.6% in those aged >85 years using the European Working Group on Sarcopenia in Older People definition. Sarcopenia is associated not only with aging, but also with various metabolic diseases, including type 2 diabetes mellitus (T2DM), fatty liver/steatohepatitis, degenerative cranial nerve disease, and inflammatory disease [2]. The key etiology of increased sarcopenia in patients with metabolic diseases is decreased mitochondrial function. In support of other studies showing that mitochondrial dysfunction is the major cause of sarcopenia [3,4], in 2019, a key factor in cancer-induced sarcopenia was identified in the regulation of mitochondrial function (Fig. 1) [5].
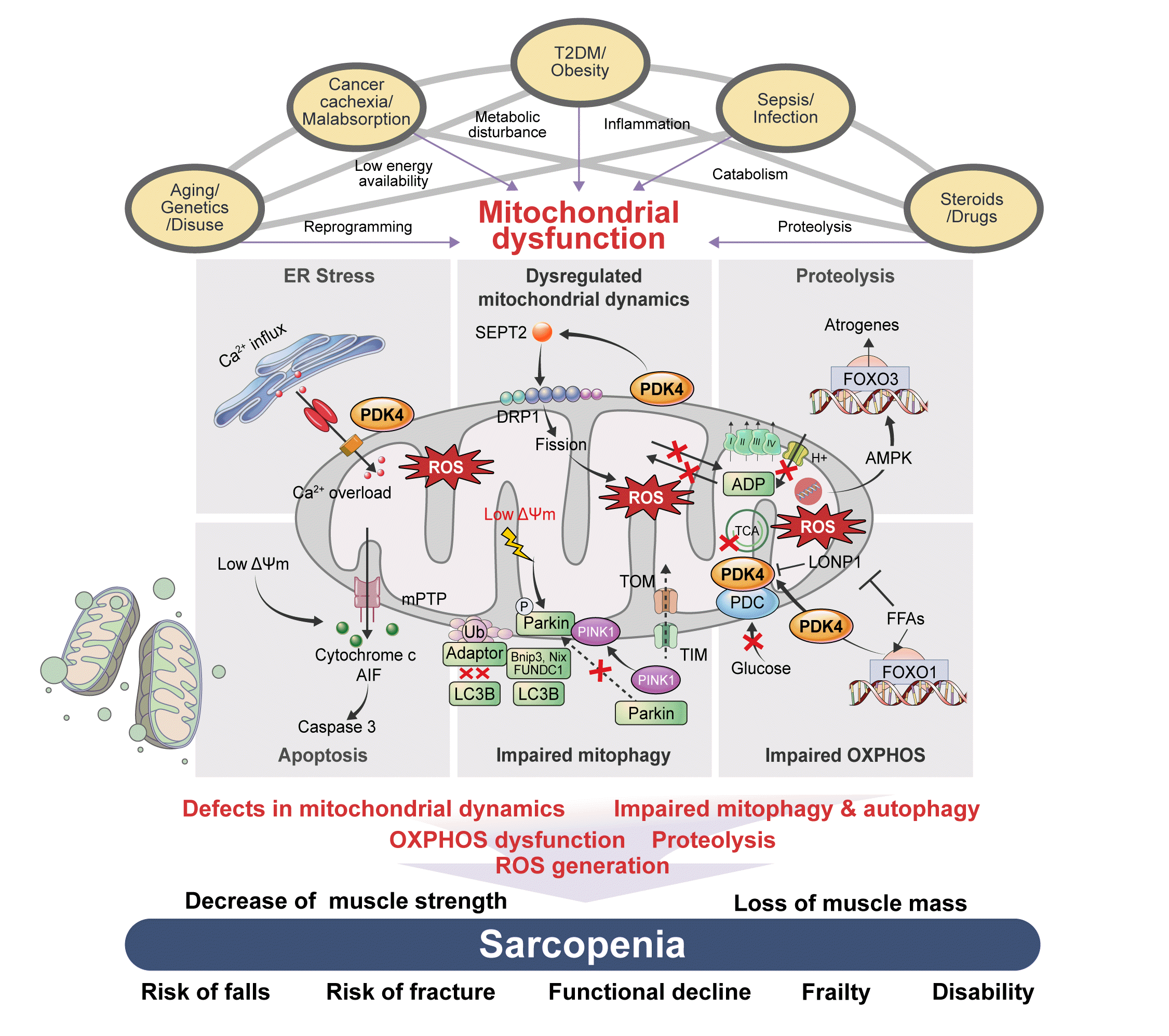
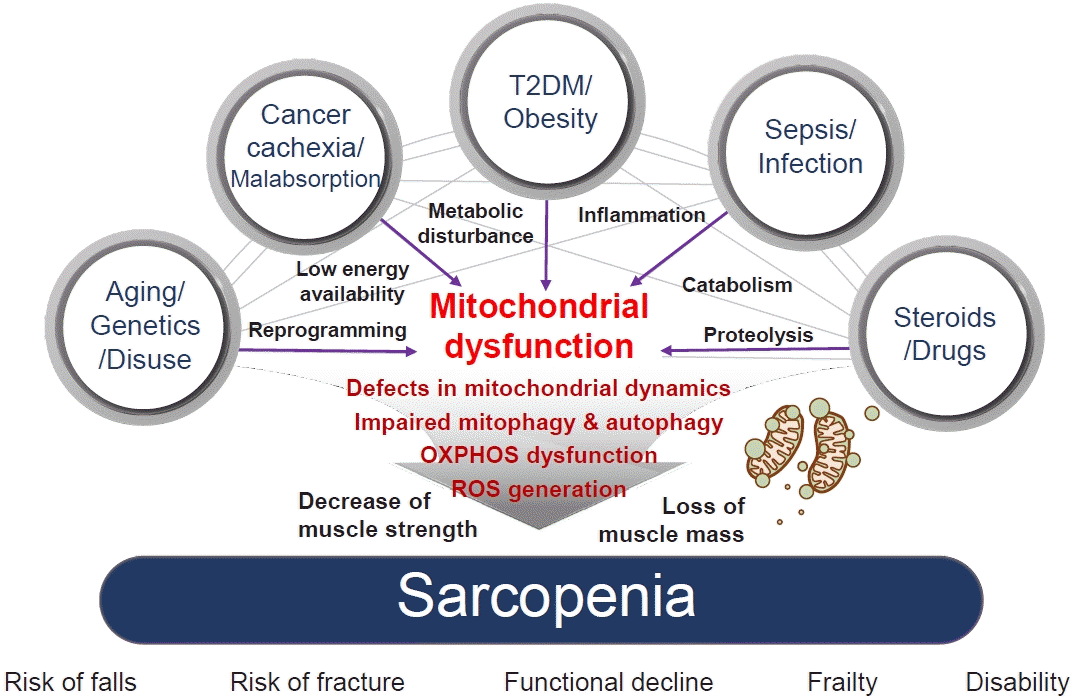 | Fig. 1.The association between risk factors of sarcopenia and mitochondrial dysfunction. Sarcopenia, generally defined as aging-related loss of muscle mass and function, is closely associated with other genetic or environmental factors. Sarcopenia pathogenesis occurs in multiple ways: genetic predisposition, aging, and disuse of muscle lead to transcriptional reprogramming or epigenetic modification; cancer cachexia or nutrient malabsorption cause low energy availability; metabolic diseases, such as type 2 diabetes mellitus (T2DM) and obesity, lead to metabolic disturbances; infection and sepsis cause inflammation; and steroids and other drugs can lead to catabolism and proteolysis. All these risk factors cause mitochondrial dysfunction, which results in sarcopenia via several mechanisms: defects in mitochondrial dynamics, such as fusion/fission; impaired mitochondrial quality control, including mitochondrial biogenesis; mitophagy and autophagy; dysfunctions in oxidative phosphorylation (OXPHOS), which is important in energy generation; and reactive oxygen species (ROS) production, which originates in dysregulated OXPHOS. Mechanistically, these factors decrease muscle strength and cause loss of muscle mass, which are associated with an increased risk of falls and fractures, functional decline, frailty, disability, and hospitalization with poorer health outcomes.
|
Mitochondria are the powerhouse of cells and are vital in maintaining muscle mass and activity [6]. Among muscle cell types, a myofiber is a postmitotic, highly differentiated cell containing mitochondria as the sole provider of energy for movement and metabolism [7]. Interestingly, the function of muscle mitochondria depends on its locality and biochemical specializations: while intermyofibrillar mitochondria are involved in oxidative phosphorylation and modulation of Ca2+ flux, subsarcolemmal mitochondria control gene expression, and reactive oxygen species (ROS) levels [8,9]. To meet the high energy demands of skeletal muscle, mitochondria rely on dynamic regulation of quality control processes, which include mitochondrial oxidant-scavenging systems, mtDNA maintenance, calcium homeostasis, protein degradation and repair, mitodynamics, and autophagy pathways. Failure to maintain these processes, as in denervation or aging, results in muscle dysfunction, making mitochondrial impairment the hallmark of various myopathies [10].
Pyruvate dehydrogenase kinases (PDKs) are a family of mitochondrial enzymes that act to suppress the conversion of pyruvate to acetyl coenzyme A via inhibitory phosphorylation of the pyruvate dehydrogenase complex (PDC) [11]. In mammals, there are four isoenzymes of PDK (PDK1, 2, 3, and 4), which are expressed in a tissue-specific manner [12]. Among all PDKs, PDK4 is one of the most well-studied because its expression is dramatically changed in different physiological conditions [13,14]. Prolonged upregulation of PDK4 is also found in mitochondrial dysfunction-related metabolic diseases, especially pathologic muscle conditions associated with metabolic disease [15]. Here, we discuss the molecular mechanisms and effects of mitochondrial dysfunction in sarcopenia, and specifically the role of PDK4 activity, and show that targeting mitochondria could be a therapeutic target for treating sarcopenia.
Go to : 
PDK4 IN MUSCLE-RELATED MITOCHONDRIAL DYSFUNCTION
Optimal mitochondrial activity depends on metabolic flexibility between processes catalyzed by various enzymes, especially the PDC, which controls pyruvate entry into the tricarboxylic acid cycle and thus the provision of adenosine triphosphate (ATP) for normal cellular activities. PDC activity is controlled by mitochondrial PDKs; among these, PDK4 is highly expressed in both starved and obese muscle tissue in animal models [16]. PDK4 regulates activity of the complex by inhibitory phosphorylation, which reduces carbon influx and, consequently, the onset of mitochondrial-mediated direct and indirect muscle pathologies. These can include muscle atrophy, sarcopenia, cancer cachexia, obesity-induced muscle insulin resistance, diabetes, and aging [5,17-20]. Moreover, PDK4 causes mitochondrial defects by interacting with proteins other than PDC, meaning the effect of PDK4 on mitochondria occurs via a range of both canonical and non-canonical pathways [21,22].
Mitochondrial energetics and PDK4 in muscle
Skeletal muscle activity relies on dynamic mitochondrial ATP production that responds to that variation in metabolic rate between rest and work. Consequently, elevations in mitochondrial ROS become a normal part of muscle physiology [23]. However, altered mitochondrial energetics and other cellular stressors can cause abnormal elevations in ROS levels. In turn, this exacerbates mitochondrial dysfunction via enhanced proteolytic degradation (autophagy); impaired mammalian target of rapamycin activity, which is required for protein synthesis; reduced ATP levels; and enhanced fission. As a result, mitochondrial dysfunction is associated with myopathies such as sarcopenia, muscle atrophy, amyotrophic lateral sclerosis, cancer cachexia, diabetes, and aging [24,25].
PDK4 activity can both affect and be affected by ROS generation. For example, oxidative skeletal muscle from genetically obese and diabetic mice shows forkhead box protein O1 (FOXO1) and peroxisome proliferator-activated receptor (PPAR)α/δ-mediated increases in PDK4 levels. This results in a metabolic shift from glucose to fatty acid oxidation, and a subsequent reduction in oxidative phosphorylation activity and increase in proton leakage, which leads to higher mitochondrial ROS levels and susceptibility to insulin resistance [26]. In addition, administration of the FOXO1-selective inhibitor AS1842856 in diabetic rat hearts improved glucose oxidation, with a concomitant decrease in PDK4 and carnitine palmitoyltransferase 1 expression, thus reducing ROS levels and improving muscle growth, suggesting that PDK4 may mediate the associations between mitochondrial dysfunction and muscle wasting (Fig. 2) [27].
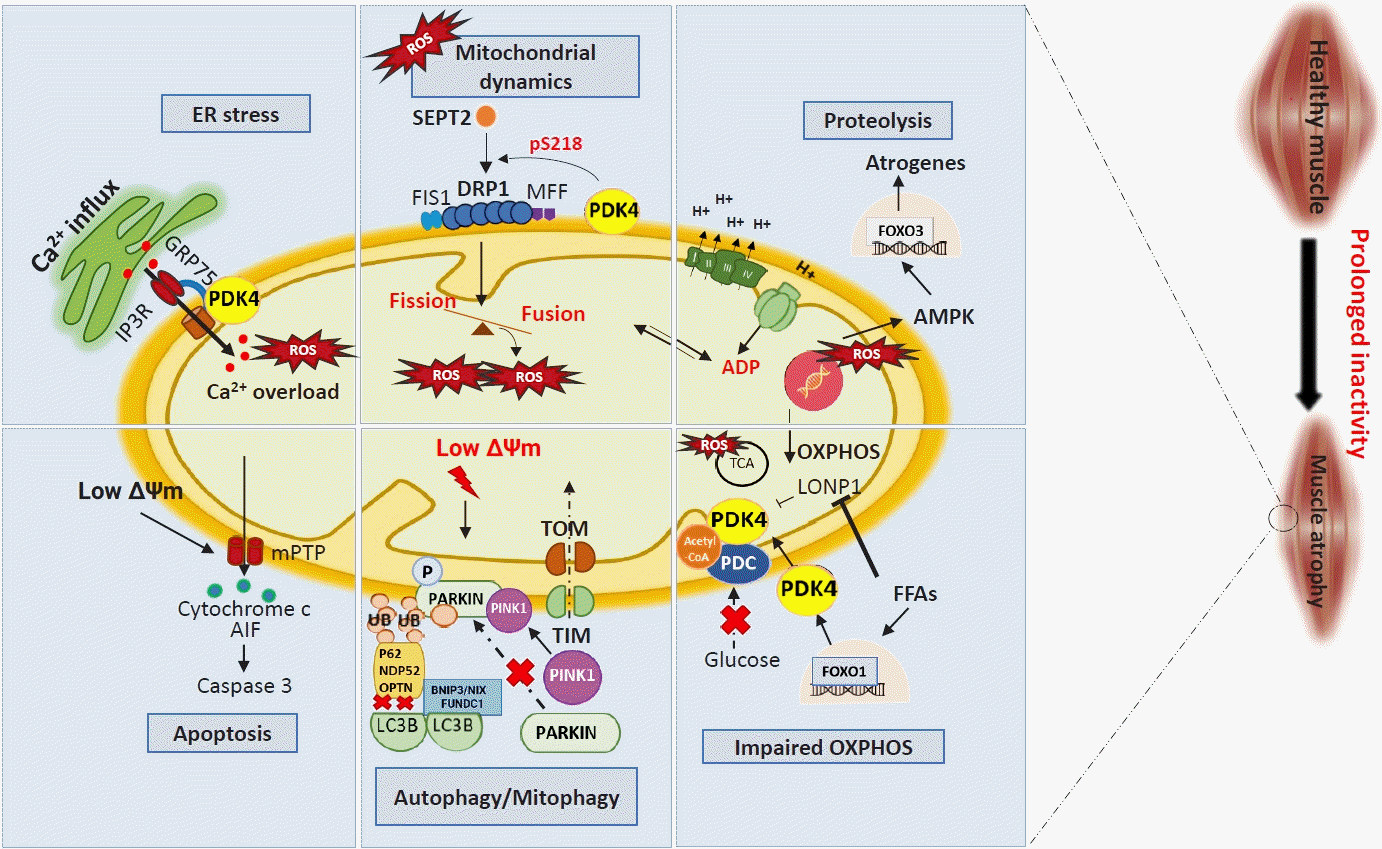 | Fig. 2.Muscle atrophy induces mitochondrial dysfunction through multiple pathways. Prolonged inactivity and other myopathies elevate reactive oxygen species (ROS) levels, which regulate different mitochondrial systems. Increased endoplasmic reticulum (ER) stress caused by calcium (Ca2+) overload and ROS results in Ca2+ influx into mitochondria through pyruvate dehydrogenase kinase 4 (PDK4)-mediated stabilization of inositol 1,4,5 trisphosphate receptor type 1 (IP3R1)–glucose-regulated protein 75 (GRP75)–voltage-dependent anion-selective channel 1 complex at the mitochondria-associated ER membrane. Increased mitochondrial Ca2+ content causes a drop in mitochondrial membrane potential (ΔΨm) leading to the opening of mitochondrial permeability transition pores (mPTP) to release cytochrome c, which activates apoptosis-inducing factor (AIF), leading to the onset of caspase 3-mediated cell death. Muscle atrophy causes increased fission via septin 2 (SEPT2)-mediated dynamin-related protein 1 (DRP1) upregulation. This imbalance between fusion and fission generates ROS and a lower ΔΨm, leading to dysfunctional mitochondrial-mediated autophagy (mitophagy). This occurs via suppressed PTEN-induced kinase 1 (PINK1)-PARKIN interaction, as well as reduced autophagic flux by lysosome adapter accumulation. Fission also causes a drop in ATP synthesis and impaired electron transport chain activity, leading to the activation of 5´ AMP-activated protein kinase (AMPK)–forkhead box protein O3 (FOXO3)-dependent atrogenes, which cause protein degradation (proteolysis). An increase in free-fatty acids (FFAs) caused by myopathologic metabolic alterations results in a switch from glucose oxidation to beta-oxidation. This causes an increase in FOXO1-PDK4 activity leading to the inactivation of the pyruvate dehydrogenase complex (PDC) by PDK4. Synergistically, FFAs also inhibit Lon peptidase 1 (LonP1), which degrades PDK4 in the mitochondria. Overall, this decreases oxidative phosphorylation (OXPHOS), leading to metabolic inflexibility-related myopathies. Acetyl-CoA, acetyl coenzyme A; ADP, adenosine diphosphate; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; FIS1, mitochondrial fission 1 protein; FUNDC1, FUN14 domain containing 1; H+, proton ion; LC3B, microtubule-associated proteins 1A/1B light chain 3B; MFF, mitochondrial fission factor; NDP52, nuclear dot protein 52; OPTN, optineurin; P62, sequestosome 1 (SQSTM1); TCA, tricarboxylic acid cycle; TIM, translocase of the inner membrane; TOM, translocase of the outer membrane; Ub, ubiquitin.
|
PDK4 and mitochondrial turnover in muscle
Mitochondrial surveillance is crucial for proper functioning and is done by specific proteases through selective removal of damaged mitochondria. Lon peptidase 1 (LONP1) is an important protease that resides in the mitochondrial matrix and is involved in muscle mitochondrial quality control. A recent study showed skeletal muscle-specific ablation of LONP1 (LONP1 mKO) resulted in impaired mitochondrial protein turnover and increased autophagy upon muscle disuse, worsening muscle loss [28]. Inactivity-induced muscle atrophy results in lower ATP output and an increase in adenosine monophosphate (AMP); this energy imbalance activates AMP-activated protein kinase, an energy sensor. In turn, this activates FOXO3, thereby increasing expression of atrogenes involved in both the ubiquitin-proteasome system and autophagy, leading to proteolysis (Fig. 2) [29]. Interestingly, in LONP1 mKO mice, comparative analysis of MitoCarta2.0 and mass spectrometry proteomics of LONP1 mKO-isolated mitochondria showed upregulation of various mitochondrial matrix proteins, including PDK4 [28]. In cardiomyocytes, PDK4 is a direct substrate of LONP1, with PDK4 turnover via this pathway regulating PDC activity [30]. Taken together, these results suggest that the effect of LONP1 on mitochondrial turnover may be mediated by PDK4.
PDK4-mediated mitochondrial-associated membrane formation in muscle
Calcium (Ca2+) is essential for muscle contraction, since it maintains mitochondrial activity; specifically, Ca2+ stimulates pyruvate dehydrogenase phosphatase 1, which dephosphorylates and activates the PDC to promote glucose metabolism. Furthermore, Ca2+ stimulates tricarboxylic acid cycle enzymes and ATP synthase to enhance ATP synthesis [31]. Sarcoplasmic reticulum is a special form of endoplasmic reticulum (ER) in striated muscle that interacts with mitochondria to exchange phospholipids and Ca2+. This inter-organelle interaction is termed a mitochondrial-associated membrane (MAM) and modulates Ca2+ transfer via formation of a macromolecular complex consisting of inositol 1,4,5-trisphosphate receptor type 1, glucose-regulated protein 75, and voltage-dependent anion channel 1. MAM-related muscle impairment is implicated in muscle dystrophy, aging, and insulin resistance, where a reduction in MAM formation aggravates these disorders [22,32].
PDK4 was recently identified in the MAM fraction, where it interacts with and stabilizes the inositol 1,4,5-trisphosphate receptor type 1–glucose-regulated protein 75-voltage-dependent anion channel 1 complex in skeletal muscle. Under normal conditions, palmitate treatment causes mitochondrial Ca2+ overload and leads to elevated ROS levels, resulting in the opening of mitochondrial permeability transition pores, cytochrome c release, and apoptosis. Moreover, MAM impairment causes ER stress and activates c-Jun N-terminal kinase and Ser/Thr-phosphorylation of insulin receptor substrate 1, resulting in insulin resistance (Fig. 2) [22,33]. However, in high-fat diet-challenged Pdk4−/− mice, MAM-related mitochondrial Ca2+ uptake and activation of PDC were reduced, resulting in enhanced glucose oxidation and improved glucose tolerance and insulin sensitivity resistance [22,33]. PDK4 therefore appears to have a vital role in the effect of fatty acid availability on MAM dysfunction and consequent metabolic perturbations.
Go to :
MITOCHONDRIAL QUALITY CONTROL AND PDK4 IN MUSCLE
As mitochondria divide frequently, a dynamic balance between fusion and elongation is required to maintain structural and functional integrity. This is done by various proteins located inside and outside the mitochondria. Key proteins that promote mitochondrial fission include dynamin-related protein 1 (DRP1) and fission 1, whereas proteins regulating mitochondrial fusion include optic atrophy 1 and mitofusion 1 and 2 (MFN1/2). Increased mitochondrial fission during prolonged inactivity is associated with muscle atrophy, with the resultant mitochondria more fragmented/round, producing more ROS, and unable to generate ATP [34]. Muscle from patients with chronic kidney disease shows mitochondrial dysfunction, with increased PDK4-mediated PDC inactivation and distorted dynamics resulting from suppressed Mfn-1 expression and a low phospho-DRP1(S637)/DRP1 ratio [35]. Various adapters interact with DRP1; however, non-canonical regulation of mitochondrial dynamics by PDK4 was recently reported, which promoted Drp1-mediated fission under stress conditions via phosphorylation of Septin2 at Serine218. This further strengthens the evidence that PDK4 directly regulates enzymes involved in mitochondrial quality processes (Fig. 2) [36].
Mitochondrial fission precedes mitophagy, which is a quality control mechanism to remove dysfunctional mitochondria as a whole or cause budding off as mitochondrial-derived vesicles. Mitophagy occurs through both ubiquitin-dependent (i.e., PTEN-induced kinase 1 [PINK1]-PARKIN) and ubiquitin-independent (e.g., BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 [BNIP3]/BNIP3L, FUN14 domain containing 1 [FUNDC1], BCL213) pathways [37]. Loss of mitochondrial membrane potential activates mitophagy and causes lysosome-associated mitochondrial degradation. Impaired mitophagy is evident in skeletal muscle during aging and sarcopenia. A cross-sectional study reported that inactive older women show significantly lower gene expression (approximately 30% to 40%) of DRP1, BNIP3, BECLIN1, autophagy related 7 (ATG7), and PARKIN in muscle [38]. Muscle-specific ablation of PARKIN in mice results in lower mitophagy flux in response to acute exercise and training; by contrast, overexpression of PARKIN in older mice inhibits mitochondrial permeability transition pore opening, thus improving muscle strength by increasing mitochondrial content [39]. A recent study showed that Mfn2 deficiency-related sarcopenia in aged muscle activates hypoxia inducible factor 1 subunit alpha (HIF1α)-BNIP3, which appears to be an adaptive mitophagy strategy to compensate for muscle loss. PDK4 is a target gene of HIF1α, and muscle from old control and young and old Mfn2-knockout mice showed increased Pdk4 mRNA levels but not Pdk1 levels [40]. In addition, PDK4 stimulates mitophagy via PINK-PARKIN stabilization by elevating pyruvate [41]. Overall, although there is some evidence that PDK4 regulates mitophagy, further studies are required.
Go to :
PDK4 IN THE REGULATION OF MUSCLE PROTEIN DEGRADATION AND ATROPHIC GENE EXPRESSION
PDK4 is expressed in all skeletal muscle types (fast-twitch glycolytic, fast-twitch oxidative, and slow-twitch oxidative), with its expression further induced by fasting [42]. The mechanism responsible for the regulation of PDK4 gene expression has been extensively studied. The transcription factors FOXO1 [43], PPARα [44], PPARδ, estrogen-related receptor-α, and transcriptional co-activator PPARγ co-activator-1 [45] are the primary regulators proposed to upregulate PDK4. In the fed condition, increased serum insulin levels activate protein kinase B, also known as AKT, leading to suppression of FOXO1-mediated PDK4 expression, whereas during starvation, a decline in insulin levels reactivates FOXO1 and enhances PDK4 expression [46]. In healthy states, this dynamic change in PDK4 levels is critical for the metabolic switch from glucose to fatty acid oxidation [11,47], which is facilitated by PDK4 having a very short half-life (approximately 1 hour) relative to other mitochondrial proteins due to its rapid degradation by the mitochondrial Lon protease [30].
There is convincing evidence that PDK4 is closely related to metabolic dysfunction and muscular atrophy. In genetically obese, diet-induced obese, and insulin-resistant mice, as well as in diabetic rats, PDK4 expression is consistently elevated in skeletal muscle, while expression of the other PDKs remain unchanged [16,22,48]. Furthermore, loss of the fatty acid translocase cluster of differentiation 36 dampens starvation-induced FOXO1 and PDK4 expression [26], suggesting that fatty acid uptake stimulates PDK4 expression. Increased expression, coupled with enhanced stability by fatty acids, prolongs upregulation of PDK4 in insulin-resistant states [16]. Indeed, that PDK4 may have a causal role in insulin resistance is suggested by previous studies showing that genetic ablation of Pdk4 protects mice from high-fat diet-induced glucose intolerance and insulin resistance [49,50], although recent studies also suggest actions of PDK2 [51] and PDK4 [52-54] that are independent of PDC regulation.
Furthermore, increased PDK4 expression correlates with aging [40], atrophic muscle [5,25], and severe muscle loss in cancer cachexia [5], T2DM [46], glucocorticoids [55], sepsis [56], amyotrophic lateral sclerosis [25], and muscle disuse [28]. Treatment of C2C12 myotubes with culture medium collected from mouse colorectal cancer cells (C26) induces PDK4 expression and markedly reduces the size of C2C12 muscle fibers, while knockdown of PDK4 prevents tumor media-induced shrinkage of muscle fiber. Furthermore, overexpression of PDK4 induces mitochondrial dysfunction and muscle fiber shrinkage [5]. Together, these findings suggest that PDK4 plays a central role in mediating metabolic instability-induced skeletal muscle dysfunction (Fig. 3).
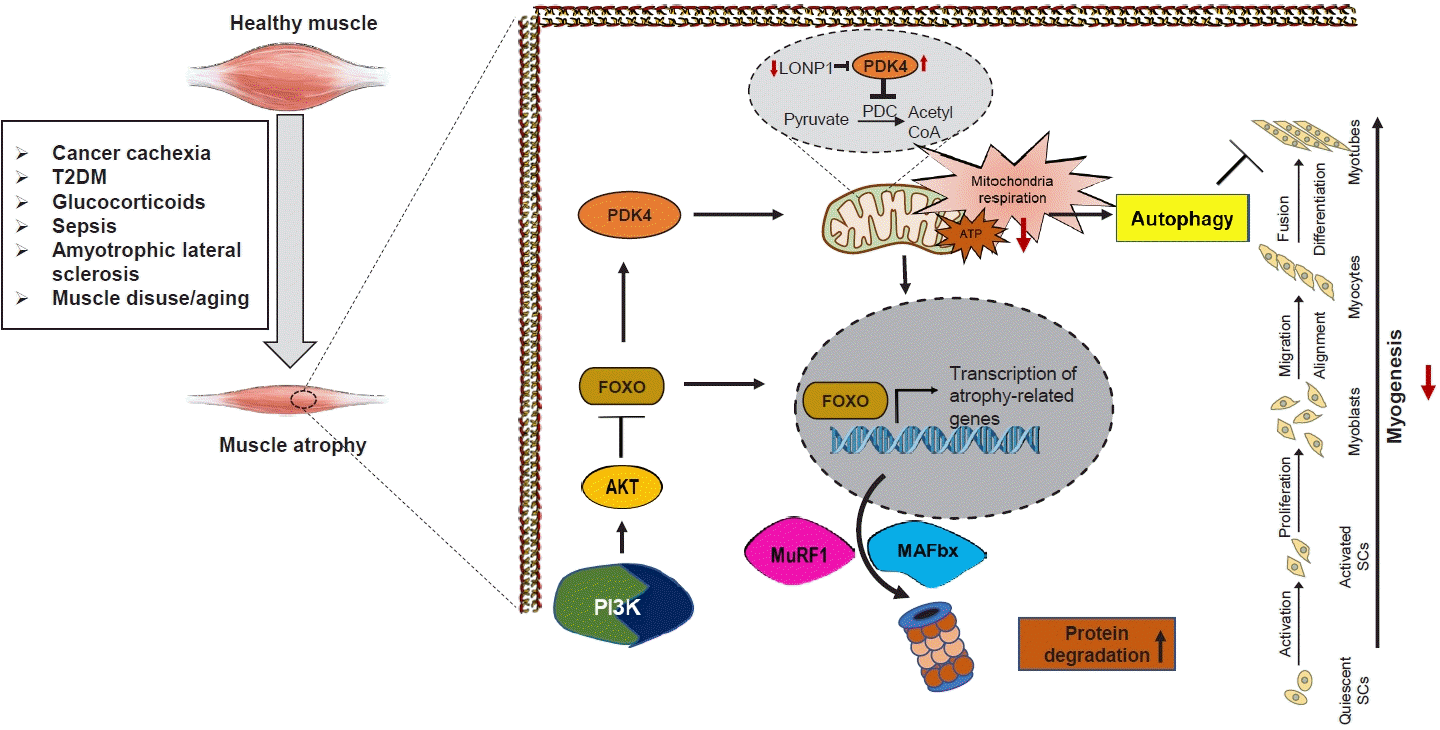 | Fig. 3.Schematic of the involvement of pyruvate dehydrogenase kinase 4 (PDK4) in regulating muscle protein degradation and atrophic gene expression. In the pathological state, PDK4 is activated and can contribute to mitochondrial damage, as well as transcription of atrophy-related genes culminating in muscle atrophy. The transcription factor forkhead box protein O1 (FOXO1) is proposed to upregulate transcription of PDK4. During starvation, a decline in insulin levels activates FOXO1, which enhances PDK4 expression. PDK4 is highly expressed in cancer cachexia, type 2 diabetes mellitus (T2DM), glucocorticoid-induced myopathy, sepsis, amyotrophic lateral sclerosis (ALS), muscle disuse, and aging. Overexpression of PDK4 induces mitochondrial dysfunction and muscle protein degradation, giving PDK4 a central role in mediating metabolic instability-induced skeletal muscle dysfunction. Acetyl-CoA, acetyl coenzyme A; ATP, adenosine triphosphate; LonP1, Lon peptidase 1; MAFbx, muscle atrophy F-box; MuRF1, muscle RING finger 1; PDC, pyruvate dehydrogenase complex; PI3K, phosphatidylinositol-3-kinase; SC, satellite cell.
|
Go to :
THE EFFECT OF THE UBIQUITINPROTEASOME PATHWAY ON PDK4 MEDIATED MUSCLE ATROPHY
Aberrant activation of protein degradation is the primary pathway leading to muscle atrophy, with the ubiquitin-proteasome pathway serving a pivotal role in the mediation of protein degradation [57]. E3 ligases mediate protein ubiquitination; proteasomes then identify poly-ubiquitinated proteins and trigger degradation [58]. There are two muscle-specific E3 ligases: muscle RING finger 1 (MuRF1) and muscle atrophy F box (MAFbx) [59]. During muscle atrophy, MuRF1 and MAFbx are overexpressed: inhibiting the function of MuRF1 and MAFbx suppresses muscle loss, attenuating muscle atrophy [60,61]. A recent study [62] indicates that overexpression or dexamethasone (Dex)-mediated induction of PDK4 enhances interaction between myogenin (MYOG) and MAFbx, but not between MYOG and MuRF1, which suggests that PDK4 promotes MAFbx recruitment to catalyze muscle atrophy-induced MYOG degradation (Fig. 4); by contrast, Pdk4−/− mice exert resistance against glucocorticoid-induced muscle atrophy. In a Dex and cancer cachexia induced mouse model of atrophy, expression of PDK4 and MAFbx are upregulated, whereas knockdown of PDK4 reduces MAFbx expression [62]. Taken together, PDK4-dependent activation of the ubiquitin-proteasome pathway appears to underly the promotion of muscle atrophy by directly targeting the muscle-specific E3 ligase MAFbx.
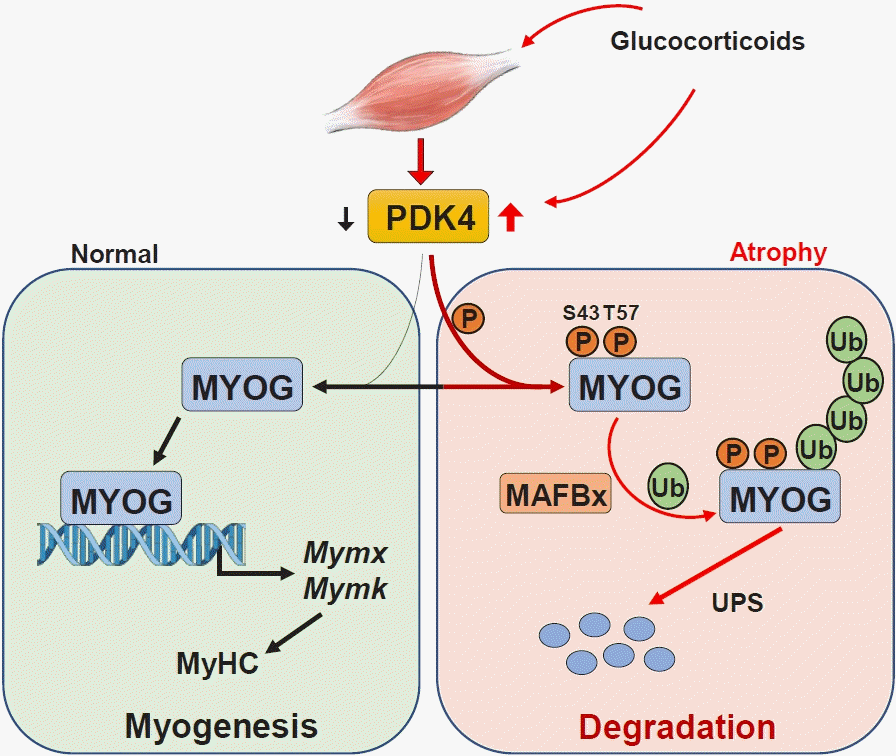 | Fig. 4.Pyruvate dehydrogenase kinase 4 (PDK4) and regulation of myogenesis. The ubiquitin-proteasome pathway serves a pivotal role in the mediation of protein degradation in muscle atrophy. In normal muscle, the transcription factor myogenin (MYOG) activates transcription of the myogenic genes MYMX (myomixer, myoblast fusion factor) and MYMK (myomaker, myoblast fusion factor). By contrast, during dexamethasone-induced muscle atrophy, induction of PDK4 leads to phosphorylation (P) of myogenin (MYOG) and recruitment of muscle atrophy F-box (MAFbx), which polyubiquitinates (Ub) MYOG, leading to its degradation and preventing transcription of myogenic genes. MyHC, myosin heavy chain; UPS, ubiquitin-proteasome system.
|
Go to :
THE MEDIATION BY PDK4 ON MUSCLE ATROPHY VIA REGULATION OF MYOGENESIS
In addition to enhanced muscle proteolysis, aberrant myogenesis is also a critical factor in muscle atrophy. Many pathological conditions such as obesity, T2DM, Cushing’s syndrome, long-term corticosteroid therapy, and cancer cachexia impair myogenesis [5,63-65]. Inactivation of myogenesis is also observed in diseases such as motor neuron degeneration, Duchenne muscular dystrophy, and spinal and bulbar muscular atrophy [66,67]. Muscle satellite cells are stem cells with self-renewal and differentiation potency, and when muscle disruption occurs, proliferative satellite cells can differentiate into myotubes and contribute to muscle regeneration [68], while defects in muscle regeneration result in muscle atrophy. Muscle regeneration is controlled by four highly related basic-helixloop-helix transcription factors known as the myogenic regulatory factors: myogenic factor 5, myoblast determination protein 1, myogenin, and myogenic regulatory factor 4/myogenic factor 6 [69].
PDK4 is implicated in the regulation of myogenesis and muscle atrophy (Fig. 4) [62]: PDK4 directly phosphorylates MYOG leading to MYOG ubiquitination and degradation. MYOG plays a crucial role in skeletal muscle differentiation and development by regulating expression of Mymk (myomaker, myoblast fusion factor), Mymx (myomixer, myoblast fusion factor), and myosin heavy chain, the key factors involved in the muscle differentiation and fusion process; as a result, suppressing PDK4 stabilizes MYOG and enhances muscle regeneration [62], while overexpression or induction of PDK4 by Dex decreases MYOG protein levels [62], suppressing myoblast differentiation and inhibiting myogenesis [5]. Thus, PDK4 has critical roles in regulating myogenic differentiation and muscle regeneration.
Go to :
CONCLUSIONS
Mitochondria are the powerhouse of cells and serve vital functions to maintain muscle mass and activity. However, mitochondria are also actively involved in metabolic changes, which can occur via MAM formation, mitochondrial quality control, and myogenesis. Evidence suggests that mitochondrial dysfunction is observed in muscle atrophy associated with metabolic syndromes; therefore, targeting mitochondrial dysfunction could be a promising therapeutic strategy for treating sarcopenia. In this review, we have discussed the recent evidence and the potential mechanisms by which PDK4 may act in mitochondrial dysfunction in sarcopenia. In addition, we discussed non-canonical pathways of PDK4-mediated muscle atrophy, which occur independently of the PDC. Our group has shown that PDK4 has non-canonical roles in MAM formation [22] and mitochondrial quality control [36], suggesting further investigation is needed to uncover other mechanisms of PDK4 involvement in regulating mitophagy and myogenesis and their potential impact on metabolic disease-related sarcopenia.
Go to :
 XML Download
XML Download