

 PDF
PDF Citation
Citation Print
Print



Introduction
Rheumatoid arthritis (RA) is a common autoimmune disorder that affects approximately 1% of the global population. It is characterized by tumor-like expansion of the synovium, angiogenesis, and destruction of the articular cartilage and bone [1]. Despite the advent of anticytokine therapies that ameliorate the inflammatory manifestations of RA, there is no curative treatment and the pathogenesis of the disease is not fully understood. In RA joints, various cell populations, including innate immune cells, adaptive immune cells, endothelial cells, and fibroblast-like synoviocytes (FLSs), are activated [1]. Resident synoviocytes participate in the chronic inflammatory response and joint destruction in RA, and in fact, these cells represent the major cell population in the invasive pannus [2]. RA-FLSs have the potential to secrete matrix-degrading enzymes (matrix metalloproteinase 3 [MMP3] and MMP9), pro-inflammatory cytokines (interleukin 1 [IL-1] and IL-6), chemokines (IL-8 and C-C motif chemokine ligand 2), and angiogenic factors (vascular endothelial growth factor and placental growth factor) [2]. Moreover, although RA synoviocytes are primary cells, they proliferate abnormally and display resistance to Fas-mediated apoptosis, similar to cancer cells [2,3]. During RA development, the phenotype of RA-FLSs is altered to an invasive and aggressive behavior, with increased migratory ability and reduced attachment-dependent growth, leading to articular cartilage destruction [2,4]. RA-FLSs can spread by migrating from the affected site to distant unaffected joints in immune-deficient mice [5]. In this regard, antimigratory agents targeting RA synoviocytes may be of therapeutic benefit. Despite the importance of FLSs in RA pathogenesis, no attempt has been made to develop an animal model capable of specifically identifying FLS migration and invasiveness. This review summarizes the immunological properties of mouse models of RA and describes humanized models that are capable of measuring the migration and invasiveness of human synoviocytes.
Ethical statements: This study was approved by the Institutional Animal Care and Use Committee (IACUC) of the Catholic University of Korea (IACUC No: CUMC-2020-0182) and the Institutional Review Board (IRB) of the Catholic Medical Center, the Catholic University of Korea (IRB No: KC14TASI0898).
Go to : 
Murine models of rheumatoid arthritis
As RA is an autoimmune disease characterized by joint destruction, careful consideration is needed when choosing the correct animal model for different in vivo experiments. Therefore, careful analysis of specific aspects of the disease and specific knowledge targeted in each study must be considered when choosing an RA animal model. Animal models of RA can be broadly divided into induced and spontaneous models, including mutant and genetically altered strains. The induced models may be polyarthritic (systemic response), which is more likely to be severe, or monoarthritic (local response). Spontaneous models progress naturally and generally involve nonresolving, chronic conditions. In this study, we have summarized the involvement of immune cells and cytokines in these models (Table 1).
Table 1.
Involvement of immune cells and cytokines in mouse models of arthritis
![]()
1. Induced models of rheumatoid arthritis
RA in animals is induced by treatment with agents, such as antigens, antibodies, adjuvants, and serum. Examples of induced mouse models of RA include collagen-induced arthritis (CIA), adjuvant-induced arthritis, proteoglycan-induced arthritis (PGIA), and collagen antibody-induced arthritis (CAIA).
1) Collagen-induced arthritis
CIA is the most frequently used animal model of RA because it shares both immunological and pathological features with human RA, including symmetric joint involvement, synovitis, and cartilage and bone destruction [2,5]. It is elicited in genetically susceptible mouse strains by immunization with type II collagen (CII) emulsified in complete Freund’s adjuvant (CFA). The requirement of T and B cells in the development of CIA is clear [6]. The autoimmune response to CII is characterized by both the stimulation of CII-specific T cells and secretion of specific immunogens and autoantigens [6]. Moreover, CD4+ T cells are believed to participate in the induction phase of the disease, supporting the activation of collagen-specific B cells [6,7].
Various cytokines have been implicated in the pathogenesis of RA through time-dependent immune system activation [8]. At the time of the clinical onset of arthritis, proinflammatory mediators (tumor necrosis factor alpha [TNF-α], IL-1β, and IL-6) and anti-inflammatory mediators (IL-10, IL-1 receptor antagonist [IL-1Ra], and transforming growth factor-beta isoforms [TGF-β1, TGF-β2, and TGF-β3]) can be detected in CIA joints [8-10]. Because interferon gamma (IFN-γ) inhibits Th17 cell development, IFN-γ receptor-knockout mice develop CIA more readily [11]. IL-23 is also required for the induction of joint inflammation by mediators including IL-17 [12,13].
2) Antigen-induced arthritis
Virtually any animal species can be used to generate antigen-induced arthritis (AIA) models. In mice, ovalbumin and bovine serum albumin (BSA) are commonly used, and modified antigens (e.g., methylated antigens) can induce chronic arthritis [14]. Antigens, such as methylated BSA, are cationic substances that bind negatively to cartilage [14,15]. When antigens are injected into one or multiple joints, inflammation progresses quickly and complement is activated locally, resulting in cartilage destruction [14]. AIA is T cell-dependent in hypothymic nu/nu mice [14]. In a recent study, both IL-23p19–/– and IL-17Ra–/– mice displayed significantly milder arthritis than wild-type mice, demonstrating that AIA progression is dependent on IL-23 and IL-17Ra signaling [16,17].
3) Proteoglycan-induced arthritis
PGIA is a progressive polyarthritis characterized by symmetrical synovitis, marginal erosion, pannus formation, and synovial infiltration of immune cells [18]. It is induced by the injection of human cartilage proteoglycans (PGs) into BALB/c (H-2d) mice, which exhibit remission and exacerbation. The first and fourth injections contain PG/CFA, the second and third injections contain PG/incomplete Freund’s adjuvant (IFA), and arthritis develops on approximately day 11 and reaches maximum severity 2 to 4 weeks after the last PG injection [19]. Mice with PGIA display CD4+ T cell responses along with an antibody response to PG, and immunoglobulin G (IgG) 2a antibody levels are correlated with disease severity [18]. Although PGIA is a predominantly Th1-type arthritis model, it has been reported that IL-4 deficiency increases the severity of PGIA [20]. In addition, treatment with IL-4 or IFN deficiency suppresses PGIA [21].
4) Collagen antibody-induced arthritis
CAIA can be induced in many susceptible mouse strains, and clinical signs of arthritis generally appear a few days after antibody administration [22]. A cocktail of anti-CII monoclonal antibodies is administered to induce arthritis in the mice. Intraperitoneal injection of lipopolysaccharide increases disease severity and the incidence of arthritis; however, it is not essential for disease induction [22].
Disease initiation in CAIA models occurs mainly independently of B and T cells; however, immune cells modulate inflammation during the effector phase [23]. The main effector cells appear to be neutrophils and macrophages, which are activated by immune complexes formed in the joints via binding of CII antibodies to cartilage surfaces [23]. Typical pathological changes observed in RA, such as synovitis, pannus formation, and cartilage and bone destruction, are observed in the joints of animals with CAIA [24,25]. Although the cytokines involved in the pathogenesis of CAIA have not been studied in detail, the involvement of IL-1β, IL-6, and TNF is anticipated [24,26].
2. Genetically modified models of rheumatoid arthritis
Genetic manipulations that affect the pathogenesis of arthritis can lead to arthritis in a variety of mice.
1) Human tumor necrosis factor transgenic mice
The first TNF transgenic mouse model was generated by Keffer et al. in 1991 [27]. They generated transgenic mice expressing wild-type and 3′-modified human TNF-α transgenes [27]. In this mouse model, synovial hyperplasia and inflammatory cell infiltration were observed in the joints after 3 to 4 weeks of age, and pannus formation, cartilage destruction, and bone erosion were observed in the mice at approximately 10 weeks of age [28,29]. This model highlights the importance of TNF-α in the pathogenesis of arthritis and can be used for the development of TNF inhibitors.
2) Interleukin-1 receptor antagonist knockout mice
IL-1 is an important inflammatory cytokine produced by a variety of cell types, including activated monocytes, macrophages, fibroblasts, and synovial cells [30]. In contrast, the IL-1 inhibitor IL-1Ra regulates the activity of IL-1 as an anti-inflammatory protein that binds to the IL-1 receptor [31,32]. It has been reported that IL-1Ra knockout mice develop spontaneous arthritis mediated by the amplification of Th17-dependent inflammation [32,33]. In a BALB/c background, polyarthritis develops starting at 5 weeks of age, and by 12 weeks of age, almost all mice are affected [33]. Histopathological analysis revealed marked synovial and periarticular inflammation, with articular destruction caused by the invasion of granulation tissues, closely resembling RA in humans [32]. Moreover, high levels of antibodies against IgG, CII, and double-stranded DNA were detectable in the sera of these mice [32,33].
3. Collagen-induced versus collagen antibody-induced arthritis
The most commonly used mouse models are CIA and CAIA. CAIA bypasses the host’s generation of autoantibodies to CII; thus, it can be induced in mice that do not possess CIA-susceptible major histocompatibility complex haplotypes (H-2q and H-2r), such as BALB/c and C57BL/6 mice [22,24]. Therefore, CAIA can be induced in most mouse strains, including transgenic and knockout mice, with an arthritis incidence approaching 100% [22]. This model is ideal for screening and evaluating anti-inflammatory agents without the effects of CFA or IFA, which strongly affect the host’s immune response.
Table 2 presents the similarities and differences between the CIA and CAIA models, and a time comparison.
Table 2.
Similarities and differences between animal models of CIA and CAIA
![]()
Go to :
Humanized models of rheumatoid arthritis
Mouse models of RA have been used for decades to study the immunopathogenesis of the disease and to explore therapeutic strategies. Nevertheless, differences in the immune systems of humans and mice have limited confidence in the success of disease treatments. To overcome this issue, severe combined immunodeficiency (SCID) mice have been used to investigate human tissues in autoimmune disease [34-37].
The use of SCID mice provides a novel model for studying tissues and cells from patients with RA [5,36]. In this review, we introduce a method that can measure cell migration and the ability to destroy human cartilage by applying synovial cells isolated from patients with RA in SCID mice.
1. Model using synoviocyte migration in severe combined immunodeficiency mice
FLSs, which are components of the synovial membrane, play a critical role in RA pathogenesis through aggressive migration and matrix invasion [2,4]. A model for measuring the migration of synoviocytes to the site of inflammation was established by subcutaneous induction of inflammation with CFA in SCID mice and then injecting synoviocytes from patients with RA around the site of inflammation [38]. One day after the CFA injection, RA-FLSs labeled with fluorescent dyes were implanted intradermally into the mice at a fixed distance (1.2 cm) from the injection site (Fig. 1). Five days after implantation, skin samples were obtained from the FLS-implanted site, CFA-injected site, and a region between these two sites (sites a, c, and b in Fig. 1). The migration of synoviocytes was analyzed according to the presence or absence of human leukocyte antigen class I (+) cells in each tissue site obtained at the end of the experiment [38].
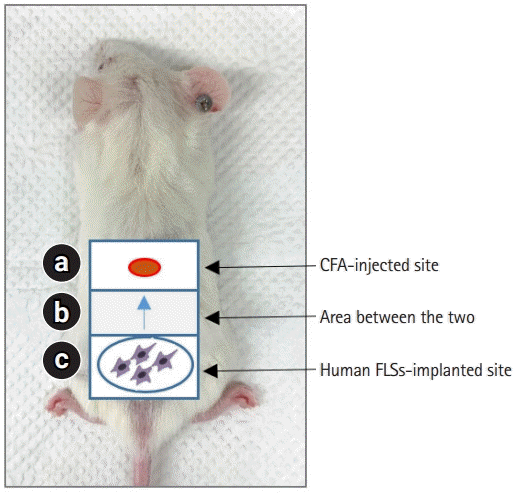 | Fig. 1.Schematic of model using synoviocyte migration in severe combined immunodeficiency mice. Skin inflammation was induced by subcutaneously injecting CFA (120 µg) into site a. One day after the CFA injection, rheumatoid arthritis-FLSs were implanted intradermally in site c. Five days after human FLSs implantation, skin samples were obtained from site b. CFA, complete Freund’s adjuvant; FLS, fibroblast-like synoviocyte.
|
2. Model using synoviocyte migration and invasion in severe combined immunodeficiency mice
RA starts in a few joints; however, as the disease progresses, it can affect all joints [5]. Interestingly, it has been reported that RA synoviocytes in a SCID mouse model can travel long distances through the bloodstream and migrate toward, attach to, and invade the distant cartilage matrix [5]. This model may be useful for elucidating the role and mechanism of an agent targeting activated synoviocytes in RA. In this section, we summarize the protocol for developing a synoviocyte migration and invasion model in SCID mice.
1) Implantation experiments
FLSs were prepared from synovial tissues of patients with RA [4,38]. Normal human cartilage was obtained from the nonarthritic knee joints of patients undergoing amputation [5,39]. Cartilage and RA synoviocytes in sponges were coimplanted into SCID mice at the primary site (6–8 weeks), and cartilage without synoviocytes was implanted at the contralateral site [5,35,39]. After 60 days, the implants were removed and either immediately embedded in a snap-frozen medium or fixed in 4% buffered formalin for paraffin embedding [5,37,39]. Using standard hematoxylin and eosin staining, each specimen was evaluated for invasion and perichondrocytic cartilage degradation in the implanted cartilage.
A procedural overview of the current model is shown in Fig. 2.
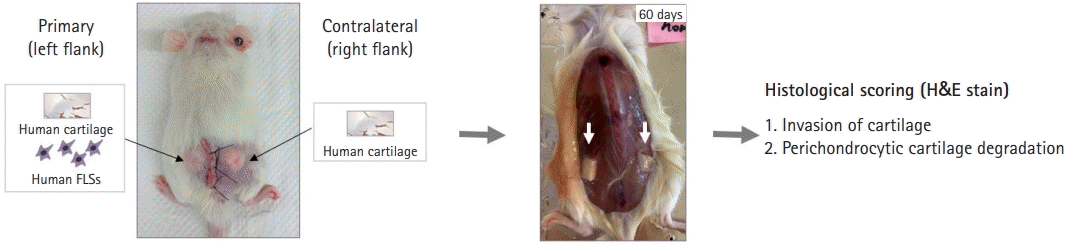 | Fig. 2.Schematic of model using synoviocyte migration and invasion SCID. Cartilage and rheumatoid arthritis-FLSs were coimplanted into SCID mice at the primary site and cartilage without cells was implanted at the contralateral site. After 60 days, the implants were removed, embedded in paraffin, and stained with hematoxylin and eosin (H&E). Implant evaluation was performed using H&E-stained sections to determine invasion and perichondrocytic cartilage degradation. SCID, severe combined immunodeficiency; FLS, fibroblast-like synoviocyte.
|
Go to :
Conclusion
The careful selection, design, and implementation of animal models are important strategies for RA treatment. In this review article, we describe a variety of mouse models that are widely used or that exhibit pathologies similar to those of arthritis in humans. These models were divided into induced and genetically modified groups. This summary of the characteristics of each model will help researchers to select the best animal model for evaluating the effectiveness of experimental treatments for RA. Furthermore, humanized models are extremely important for investigating the role and mechanism of RA-FLSs targeting these cells.
Go to :
 XML Download
XML Download