

 PDF
PDF Citation
Citation Print
Print




INTRODUCTION
Acute myocardial infarction (AMI) refers to severe myocardial ischemia caused by coronary artery occlusion and further promotes myocardial necrosis, which destroys life quality and even loses patients’ lives. Currently, ischemia-reperfusion (I/R) therapy can better improve the injury and necrosis of cardiomyocytes.1) However, I/R therapy is very time-limited and has been reported to cause secondary damage to the myocardium,2) which significantly affects the application of this method in AMI. Moreover, the mechanism of cardiomyocyte injury caused by AMI is terrifically complicated. Hence, it is essential to probe the biological mechanism of cardiomyocyte injury caused by AMI.
Ferroptosis is a new type of iron-dependent programmed cell death, different from apoptosis, cell necrosis, and autophagy. The main mechanism of ferroptosis refers to the high expression of unsaturated fatty acids on the cell membrane, which is catalyzed and is happened to lipid peroxidation (LPO) under bivalent iron or ester oxygenase, thus inducing cell death.3) Ferroptosis plays crucial roles in multiple biological processes and different diseases. As reported previously, ferroptosis inhibition in cardiomyocytes could alleviate AMI progression,4) indicating that suppressing ferroptosis might be a therapy for AMI. However, there are few studies about ferroptosis in AMI. Hence, this study aims to understand ferroptosis's role in AMI better.
Long non-coding RNAs (lncRNAs) with more than 200 nucleotides in length belong to non-coding RNAs and mediate physiological and pathological processes of various diseases, including AMI. For example, lncRNA MIAT knockdown attenuated hypoxia-induced cardiomyocyte injury.5) Of note, we used KEGG data analysis and found that lncRNA AC005332.7 (AC005332.7) expression was abnormally decreased in peripheral blood samples of AMI patients in comparison with that from healthy participants (has not been published), indicating AC005332.7 might be involved in AMI progression. However, the functions and regulatory mechanisms of AC005332.7 in AMI are still in the dark.
MicroRNA (miRNA), with a length of about 22 nucleotides, is another member of non-coding and plays indispensable roles in biological processes. MiR-331-3p was extensively investigated in numerous diseases such as Alzheimer's disease, myocardial infarction, etc.6)7) Horváth et al.7) found that miR-331-3p could serve as a biomarker in ST-segment-elevation myocardial infarction, suggesting miR-331-3p might be a critical factor in AMI. It is well known that lncRNAs could bind to miRNAs to mediate biological processes in the cardiovascular system.8)9) The Starbase website predicted that the binding site exists between AC005332.7 and miR-331-3p. Therefore, we assumed that AC005332.7 could interact with miR-331-3p to regulate AMI.
Dozens of research have explored the crucial roles of cyclin D2 (CCND2) in regulating the biological functions of cardiomyocytes.10) For instance, it was found that CCND2 overexpression could accelerate infarct regression and alleviate ischemia-induced cardiomyocyte injury.11) Of note, Ni et al.12) proposed that CCND2 was negatively correlated with ferroptosis in diabetic cardiomyopathy. Occasionally, Starbase prediction found that miR-331-3p had a potential binding site on CCND2, which has not been established in AMI. Therefore, we speculated that miR-331-3p might regulate CCND2 to mediate ferroptosis in AMI.
Based on the above evidence, we reasonably raised our hypothesis that AC005332.7 enhances CCND2 through sponging miR-331-3p, suppressing ferroptosis, and alleviating AMI, which might provide novel targets for AMI therapy.
METHODS
Ethical statement
The animal experiments were approved by the Affiliated Hospital of Guilin Medical University. The ethical number was GLMC202003024. Informed consent was obtained from study participants.
Cell culture and treatment
Human myocardial cell line AC16 cells were bought from American Type Culture Collection (Manassas, VA, USA) and were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Gibco, Grand Island, NY, USA), streptomycin (100 μg/mL), penicillin (100 U/mL), norepinephrine (0.1 mM) and L-glutamine (2 mM) (Sigma, Burlington, MA, USA) under the condition of 5% CO2 and 95% air at 37°C. After 48 hours, PBS washed AC16 cells 3 times and added a sugar-free DMEM medium was to culture AC16 cells. Cells were under the condition of 94% N2, 5% CO2, and 1% O2 airflow for 6, 12, and 24 hours, respectively.
Cell transfection
MiR-331-3p mimics (sense: 5′-GCCCCUGGGCCUAUCCUAGAA-3′, antisense: 5′-UUCUAGGAUAGGCCCAGGGGC-3′, Cat.No: B02001), inhibitor (5′-TATGGTCCCAGGGATCCCAGA-3′, Cat.No: B03001) and the short hairpin RNA of CCND2 (sh-CCND2, 5′-CGACTTTAAGTTTGCCATGTA-3′, Cat.No:C02007), as well as their negative control (NC) groups (mimics/inhibitor NC and sh-NC), were obtained from GenePharma (Shanghai, China). Lentiviruses carrying sh-AC005332.7 (5′-TGGTACTTATATGTACTAATA-3′, Cat.No: D02001) or overexpression vector of AC005332.7 (oe-AC005332.7, Cat.No: GL-02) were acquired from GenePharma. AC16 cells were seeded onto 6-well plates (1×105 cells/well) for transfection. When cells were in the state of 50% confluence, these plasmids were transfected into AC16 cells using Lipofectamine™ 3000 (Invitrogen, Waltham, MA, USA), referring to the manufacturer’s protocol. As for the infections of lentiviruses for cells, moderate viral supernatant was added to AC16 cells. After 24-hour infection, the fresh medium replaced the culture medium containing the virus to culture AC16 cells.
Quantitative reverse transcription polymerase chain reaction
TRIzol reagent (Invitrogen) was used to extract RNAs from AC16 cells and myocardial tissue of mice. Prime Script Reverse Transcription Reagent Kit (TaKaRa, Kusatsu, Japan) was applied to synthesize cDNA, and SYBR Premix Ex Taq II Kit (TaKaRa) was used to process qPCR. The primer sequences were presented as follows.
AC005332.7 (F): 5′-AACGCGCATCTTAGCTCATT-3′
AC005332.7(R): 5′-TCATTACGGATTTCGGGAAG-3′
miR-331-3p (F): 5′-GCCGAGGCCCCTGGGCCTATC-3′
miR-331-3p (R): 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATA CGACTTCTAG-3′
CCND2 (F): 5′-AGTCCCGACTCCTAAGACCC-3′
CCND2 (R): 5′-GAAGTCGTGAGGGGTGACTG-3′
U6 (F): 5′-CTCGCTTCGGCAGCACA-3′
U6 (R): 5′-AACGCTTCACGAATTTGCGT-3′
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (F): 5′-AGCCCAAGATGCCCTTCAGT-3′
GAPDH (R): 5′-CCGTGTTCCTACCCCCAATG-3′
All data were calculated by using the 2−ΔΔCt formula. GAPDH or U6 served as a reference gene.
The 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide assay
The cell viability was determined using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide (MTT) method. Shortly, AC16 cells were implanted onto 96-well plates (1×104 cells/well) and were cultured overnight. AC16 cells were subjected to corresponding transfections. Each well was added with ten μL MTT (0.5 mg/mL; Macklin, China) and incubated for 4 hours. Next, 150 μL DMSO was used to dissolve the crystal. The absorbance at 490 nm was acquired with a microplate reader (Thermo Fisher Scientific).
Enzyme-linked immunosorbent assay
We collected the cells or serum and measured the levels of creatine kinase (CK) and lactate dehydrogenase (LDH) with corresponding commercial kits (Nanjing Jiancheng Bioengineering, Nanjing, China). Glutathione (GSH) enzyme-linked immunosorbent assay kit (ml063305; Mlbio, Shanghai, China) was used to measure GSH levels in tissues or cells according to the manufacturer’s manual.
The measurement of reactive oxygen species
The reactive oxygen species (ROS) levels in AC16 cells and myocardial tissues of mice were detected using a ROS kit (Beyotime Biotechnology, Shanghai, China). Following the manufacturer’s instruction, DCFH-DA (2′, 7-dichloro-fluorescein diacetate) was incubated for 30 minutes under darkness at room temperature. Fluorescence intensity was measured using a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Fluorescence intensity was positively related to intracellular ROS levels. Image J software (National Institutes of Health, Bethesda, MD, USA) was performed to analyze image intensity.
Lipid peroxidation assay
The level of malondialdehyde (MDA) was determined using a Lipid Peroxidation Assay Kit (#ab118970; Abcam, Cambridge, UK). The MDA in the sample reacts with the 2-thiobarbituric acid (TBA) method to detect LPO content in cells and tissue homogenates supernatants. Shortly, the samples were added and mixed with TBA reagent upon the heating condition of a boiling water bath for 30 minutes. After being centrifuged, the supernatants were detected the absorbance at 532 nm.
Detection of iron content
Iron Assay Kit (#ab83366; Abcam) was employed to detect iron concentrations in AC16 cells or tissues. Briefly, supernatants of cells and homogenates of tissues were placed onto 96-well plates and then incubated with a five μL iron reducer at 25°C for 30 minutes to examine total iron content. Then, under a dark environment, a 100 μL iron probe incubated samples for 60 minutes at 25°C. Finishing these processes, the absorbance at 593 nm was detected using a microplate reader.
Western blotting
The proteins were acquired from AC16 cells or myocardium of mice with RIPA buffer (Beyotime Biotechnology). The protein concentration was determined with a BCA protein kit (Beyotime Biotechnology). The proteins were isolated in sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto the polyvinylidene difluoride (PVDF) membrane. After 5% bovine serum albumin (BSA) incubation, the membrane was incubated with primary antibodies CCND2 (1:2,000, #PA5-115077; Thermo Fisher Scientific), glutathione peroxidase 4 (GPX4, 1:3,000, #ab125066; Abcam), acyl-CoA synthetase long-chain family member 4 (ACSL4, 1:20,000, #ab155282; Abcam) and GAPDH (1:3,000, #ab8245; Abcam) overnight at 4°C. Afterward, horseradish peroxidase (HRP)-conjugated secondary antibody was used to incubate the PVDF membrane for 1 hour. ECL chemiluminescent reagent (Beyotime Biotechnology) was used to monitor protein bands. The densitometry analysis was computed by Image J.
Dual-luciferase reporter gene assay
The sequences of AC005332.7 or CCND2, which bound to miR-331-3p, were cloned into pGL3 vectors (Cat.No: E1761; Promega, Madison, WI, USA) to build reporter vectors. AC005332.7 wild type/mutant (WT/MUT) or CCND2 WT/MUT were co-transfected into AC16 cells with miR-331-3p mimics or NC (NC mimics), respectively, by lipofectamine 3000. The cells were incubated for 24 hours to determine the luciferase activity with a dual luciferase reporter assay system (Promega, Beijing, China).
RNA-binding protein immunoprecipitation
The correlation between AC005332.7 and miR-331-3p was validated using an RNA-binding protein immunoprecipitation (RIP) assay kit (Sigma). Shortly, the anti-Ago2 antibody was used to immunoprecipitate the chromatin, and immunoglobulin G (IgG) served as a control. AC005332.7 and miR-331-3p enrichment were measured using quantitative reverse transcription polymerase chain reaction (qRT-PCR).
RNA pull-down
The RNA pull-down kit (BersinBio, Guangzhou, China) was performed to verify the interplay between miR-331-3p and CCND2. The biotin-labeled probe with miR-331-3p was designed and synthesized by GenePharma. After the biotin-miR-331-3p probe was incubated with cell lysates, RNA-protein complexes were formed. Then, streptavidin-coupled magnetic beads were used to separate the complexes. Finally, qRT-PCR was employed to measure the expression of complexes.
Animals
The animal experiments were approved by the Affiliated Hospital of Guilin Medical University (ethical number: GLMC202003024). Forty male C57BL/6 mice aged 6–8 weeks were purchased from the Affiliated Hospital of Guilin Medical University. All mice were allowed to acquire feed and water freely and were raised in a pathogen-free surrounding with suitable temperature and humidity and a light/dark cycle every 12 hours. A week later, ten mice served as control groups; the other mice were accepted with the surgery of ligation of the left anterior descending (LAD). These mice were grouped into control, AMI, AMI+oe-NC, and AMI+oe-AC005332.7. For establishing the mice model of AMI, mice were accepted with the surgery of LAD referring to the previous study.13) The mice were anesthetized with 2.5% isoflurane for induction and 1.0% for maintenance. Next, to simulate the phenomenon of AMI, we adopted a 7-0 silk suture and then placed it around the LAD branch of the coronary artery, and the silk suture was knotted to prevent blood flow. The sham group mice underwent similar surgery in addition to ligature placement. Afterward, according to the former study, the AC16 cells with lentiviruses carrying oe-AC005332.7 were injected into the AMI mice in situ.14)15) After 72 hours, all mice were processed for euthanasia, and the myocardium and blood samples were collected for a subsequent experiment.
Hematoxylin and eosin staining
The myocardial tissues of mice were fixed with a 4% paraformaldehyde solution. Next, the samples were embedded in paraffin and cut into 5 µm thick serial sections. After dehydrating with gradient ethanol, the sections were stained with hematoxylin and eosin (H&E) solution (Beyotime Biotechnology). The photographs of the stained sections were obtained using an optical microscope with a camera.
Immunohistochemistry
Immunohistochemistry (IHC) was applied to determine the protein expression of Ki67, CCND2, and GPX4. Briefly, the myocardium of mice was embedded using 4% paraformaldehyde and paraffin. After repairing the antigen, the sections were blocked with 1% BSA and incubated with antibodies against Ki67 (Abcam), CCND2 (Thermo Fisher Scientific), and GPX4 (Abcam). The sections were then incubated with HRP-labelled antibodies. The images were acquired using a Nikon digital camera system (Nikon, Tokyo, Japan) combined with an Olympus microscope (Olympus, Tokyo, Japan).
Data analysis
GraphPad Prism 6.0 (GraphPad Software, Boston, MA, USA) was applied for data analyses. Data were shown as the mean ± standard deviation. All data were obtained from 3 independent experiments, each in triplicate. The statistical analysis methods with Student’s t-test or one-way analysis of variance were employed to calculate statistical differences. The normality of data was assessed by the Shapiro-Wilk test. Considering a significance level of 5%, there were no significant deviations from the normality of all data (p>0.05). p<0.05 was regarded as statistically significant.
RESULTS
Oxygen and glucose deprivation reduced AC005332.7 expression and promoted ferroptosis in AC16 cells
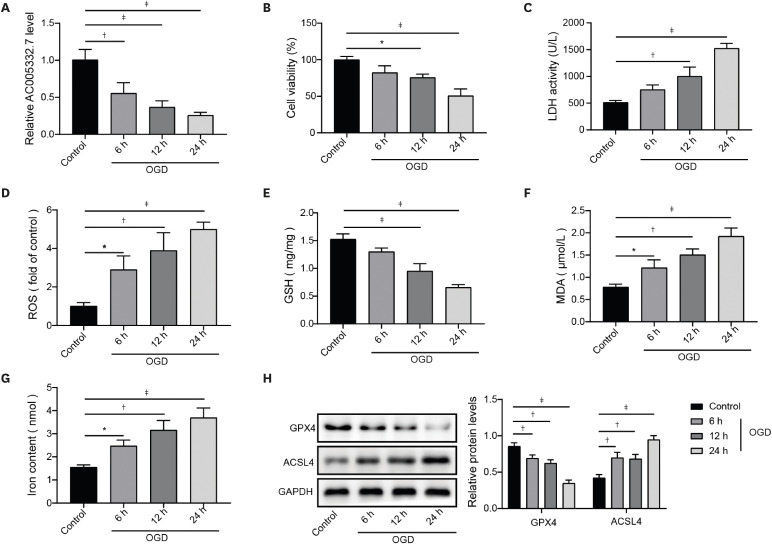
To explore whether AC005332.7 expression was affected in AMI, AC16 cells were stimulated with oxygen and glucose deprivation (OGD) for 6, 12, and 24 hours, respectively. With the extension of time in OGD exposure, AC005332.7 expression in AC16 cells was gradually reduced (Figure 1A). Additionally, cell viability of AC16 cells was dramatically impeded by OGD disposal with prolongation of time (Figure 1B). On the contrary, LDH activity gradually increased following extension with OGD exposure (Figure 1C). As for the influences of OGD stimulation on ferroptosis, we detected the levels of ferroptosis markers (ROS, GSH, MDA, iron, GPX4, and ACSL4). The results displayed that the levels of GSH and GPX4 were obviously decreased, while the levels of ROS, MDA, iron, and ACSL4 were prominently elevated with the extension of time in OGD exposure (Figure 1D-H). Based on the results above, we selected the time point of 24 hours under the OGD condition for subsequent experiments. OGD stimulation declined AC005332.7 expression and positively regulated the ferroptosis process.
Figure 1
OGD reduced AC005332.7 expression and promoted ferroptosis in AC16 cells. (A) qRT-PCR was applied to detect AC005332.7 expression. (B) MTT was used for evaluating cell viability. (C-G) LDH, ROS, GSH, MDA, and iron levels were detected using corresponding kits and methods. (H) Western blot was used for examining GPX4 and ACSL4 expression. Data were obtained from 3 independent experiments. Data were shown as mean±standard deviation (n=3).
ACSL4 = acyl-CoA synthetase long chain family member 4; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; GSH = glutathione; LDH = lactate dehydrogenase; GPX4 = glutathione peroxidase 4; MDA = malondialdehyde; MTT = 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide; OGD = oxygen and glucose deprivation; qRT-PCR = quantitative reverse transcription polymerase chain reaction; ROS = reactive oxygen species.
*p<0.05; †p<0.01; ‡p<0.001.
Overexpression of AC005332.7 repressed ferroptosis in oxygen and glucose deprivation-induced AC16 cells
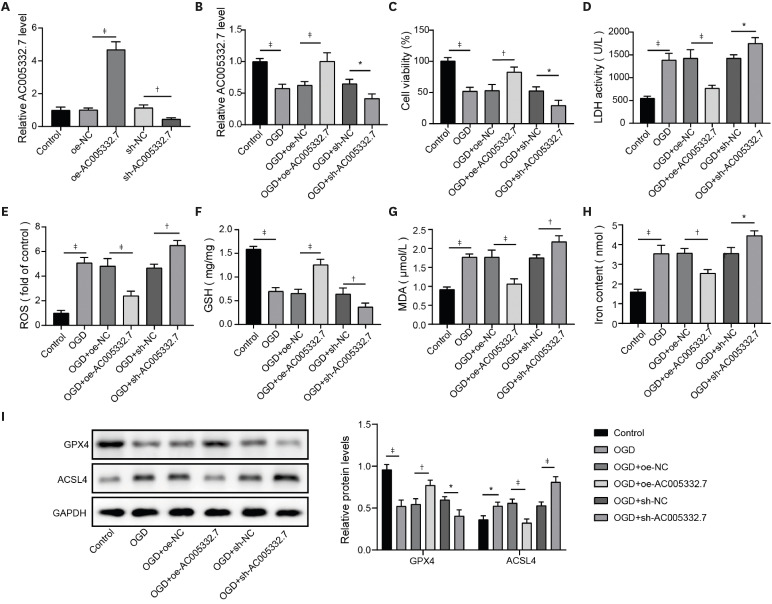
Subsequently, we probed the function of AC005332.7 on ferroptosis in OGD-induced AC16 cells. Firstly, AC005332.7 was overexpressed or silenced in AC16 cells by lentiviruses carrying oe-AC005332.7 or sh-AC005332.7, and qRT-PCR assay verified the successful transfection (Figure 2A). Then, the OGD condition decreased AC005332.7 expression, which was abolished by AC005332.7 overexpression, while further downregulated by AC005332.7 knockdown (Figure 2B). Besides, the overexpression of AC005332.7 enhanced cell viability in OGD-induced AC16 cells, and the knockdown of AC005332.7 suppressed cell viability (Figure 2C). AC005332.7 sufficiency inhibited OGD-induced LDH level, while AC005332.7 silencing aggravated the promoting effect on LDH level caused by OGD (Figure 2D). Furthermore, OGD condition trigged cell ferroptosis in which GSH and GPX4 were decreased and ROS, MDA, iron, and ACSL4 were elevated. In contrast, these phenomena were reversed by AC005332.7 overexpression but further exacerbated by AC005332.7 silencing (Figure 2E-I). These results suggested that AC005332.7 suppressed OGD-induced ferroptosis in AC16 cells.
Figure 2
AC005332.7 repressed ferroptosis in OGD-induced AC16. (A) qRT-PCR was applied to detect AC005332.7 expression. (B) qRT-PCR was used for detecting AC005332.7 expression. (C) MTT was employed for evaluating cell viability. (D-H) LDH, ROS, GSH, MDA, and iron levels were examined using corresponding kits and methods. (I) Western blot was conducted to investigate GPX4 and ACSL4 expression. Data were shown as mean±standard deviation (n=3).
ACSL4 = acyl-CoA synthetase long-chain family member 4; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; GPX4 = glutathione peroxidase 4; GSH = glutathione; LDH = lactate dehydrogenase; MDA = malondialdehyde; MTT = 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide; NC = negative control; OGD = oxygen and glucose deprivation; qRT-PCR = quantitative reverse transcription polymerase chain reaction; ROS = reactive oxygen species.
*p<0.05; †p<0.01; ‡p<0.001.
AC005332.7 indirectly mediated cyclin D2 expression through sponging miR-331-3p
As per the previous description, miR-331-3p and CCND2 were implicated in AMI.7)11) Here, we observed that OGD treatment gradually increased miR-331-3p and decreased CCND2 in a time-dependent manner (Figure 3A-C). Subsequently, miR-331-3p mimics and inhibitors were transfected into AC16 cells to overexpress and silence miR-331-3p expression, respectively (Figure 3D). In addition, the Starbase website predicted AC005332.7 had a potential binding site on miR-331-3p (Figure 3E). MiR-331-3p mimics obviously decreased luciferase activity in the WT-AC005332.7 group, but there were no such changes in the MUT-AC005332.7 group (Figure 3F). RIP assay determined that AC005332.7 and miR-331-3p were markedly enriched by Ago2 rather than IgG (Figure 3G). Moreover, miR-331-3p expression was significantly decreased by lentivirus-carrying oe-AC005332.7. Oppositely, miR-331-3p expression elevated when AC005332.7 was knocked down by sh-AC005332.7 (Figure 3H). Expectedly, OGD increased miR-331-3p expression in AC16 cells, blocked by AC005332.7 sufficiency but further promoted by AC005332.7 knockdown (Figure 3I). This evidence proved that AC005332.7 could interact with miR-331-3p and negatively regulated miR-33-3p expression.
Figure 3
AC005332.7 indirectly mediated CCND2 expression through sponging miR-331-3p. (A, B) qRT-PCR was applied for detecting miR-331-3p and CCND2 expression. (C) Western blot was conducted to examine CCND2 expression. (D) qRT-PCR was used to determine miR-331-3p expression in AC16 cells transfected with miR-331-3p mimics and inhibitors. (E) Starbase predicted the potential binding site between AC005332.7 and miR-331-3p. (F) A dual luciferase reporter gene assay was conducted to evaluate luciferase activity. (G) RIP was used to validate the interaction between AC005332.7 and miR-331-3p. (H) qRT-PCR was employed to detect miR-331-3p expression. (I) qRT-PCR was used to examine miR-331-3p expression. (J) Starbase predicted the potential binding site between miR-331-3p and CCND2. (K) A dual luciferase reporter gene assay was conducted to evaluate luciferase activity. (L) RNA-pull down was employed for validating the interaction between miR-331-3p and CCND2. (M-R) qRT-PCR and western blot were applied to measure CCND2 expression. Data were shown as mean±standard deviation (n=3).
CCND2 = cyclin D2; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; IgG = immunoglobulin G; MUT = mutant; NC = negative control; OGD = oxygen and glucose deprivation; qRT-PCR = quantitative reverse transcription polymerase chain reaction; RIP = RNA-binding protein immunoprecipitation; WT = wild type.
*p<0.05; †p<0.01; ‡p<0.001.
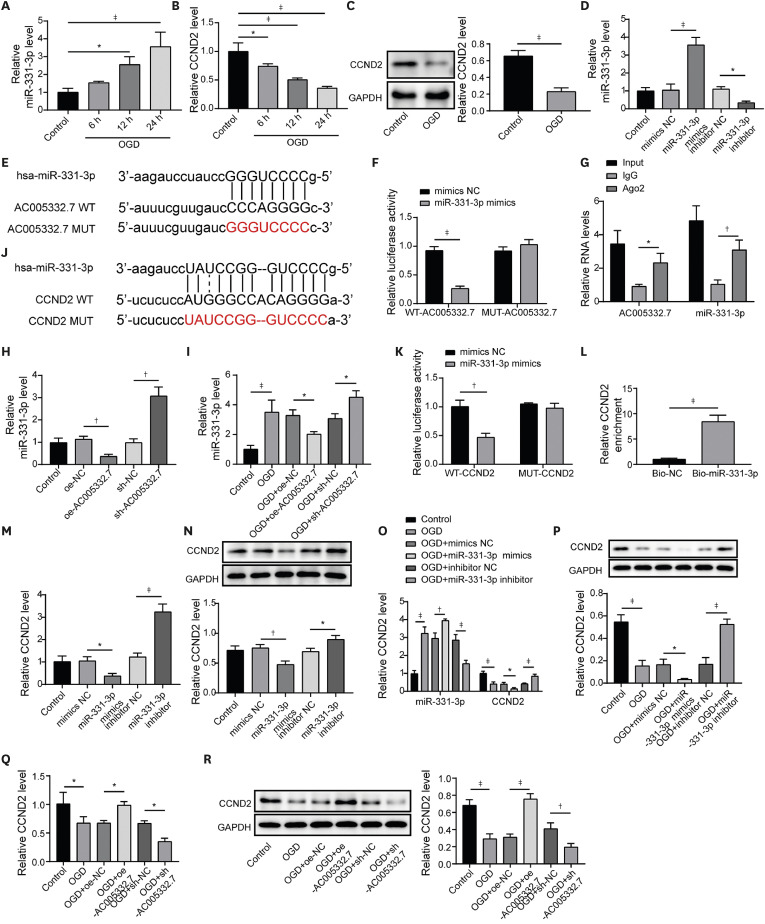
Meanwhile, we predicted the downstream molecule of miR-331-3p through the Starbase website. We found that CCND2 had a binding site to miR-331-3p (Figure 3J). Dual luciferase reporter gene assay exhibited that luciferase activity was distinctly repressed in WT-CCND2 with miR-331-3p mimics transfection, while had no influence in MUT-CCND2 with miR-331-3p mimics (Figure 3K). RIP assay revealed that CCND2 expression was obviously enriched by bio-miR-331-3p (Figure 3L). The above results indicated that miR-331-3p could target binding to CCND2. As expected, miR-331-3p mimics suppressed mRNA and protein expression of CCND2 while miR-331-3p inhibitor obtained opposite results (Figure 3M and N). OGD condition elevated miR-331-3p expression while suppressing CCND2 expression, and these results were abolished by miR-331-3p inhibitor and reinforced by miR-331-3p mimics (Figure 3O and P). Noteworthily, AC005332.7 overexpression abrogated OGD-induced inhibition of CCND2 expression, and AC005332.7 knockdown promoted OGD-induced inhibition of CCND2 expression (Figure 3Q and R). In total, our results verified that AC005332.7 negatively regulated miR-331-3p by interacting with miR-331-3p and miR-331-3p targeted to inhibit CCND2.
AC005332.7 inhibited ferroptosis in oxygen and glucose-deprivation-induced AC16 cells via reducing miR-331-3p expression
A rescue experiment was conducted to probe whether miR-331-3p is involved in AC005332.7-mediated inhibition of ferroptosis in AC16 cells upon OGD condition. The detailed groups were Control, OGD, OGD+oe-NC+mimics NC, OGD+oe-AC005332.7+mimics NC, OGD+oe-AC005332.7+miR-331-3p mimics. Firstly, miR-331-3p expression was detected by qRT-PCR. AC005332.7 upregulation inhibited miR-331-3p expression in OGD-induced AC16 cells, whereas this tendency was offset by miR-331-3p mimics (Figure 4A). MiR-331-3p elevation impaired AC005332.7 upregulation-mediated promotion of cell viability in OGD-induced AC16 cells (Figure 4B). AC005332.7 upregulation repressed LDH levels in OGD-induced AC16 cells, but miR-331-3p mimics abolished the inhibiting impact of AC005332.7 overexpression (Figure 4C). AC005332.7 overexpression increased the levels of GSH and GPX4 and decreased the levels of ROS, MDA, iron, and ACSL4 in OGD-induced AC16 cells, which these phenomena were reversed by miR-331-3p overexpression (Figure 4D-H). Therefore, we concluded that AC005332.7 inhibited ferroptosis in OGD-induced AC16 cells by inhibiting miR-331-3p.
Figure 4
AC005332.7 inhibited ferroptosis in OGD-induced AC16 cells via reducing miR-331-3p expression. (A) qRT-PCR was applied for detecting miR-331-3p expression. (B) MTT was conducted to evaluate cell viability. (C-G) The LDH, ROS, GSH, MDA, and iron levels were tested using corresponding kits and methods. (H) Western blot was applied to examine GPX4 and ACSL4 expression. Data were shown as mean±standard deviation (n=3).
ACSL4 = acyl-CoA synthetase long-chain family member 4; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; GPX4 = glutathione peroxidase 4; GSH = glutathione; LDH = lactate dehydrogenase; MDA = malondialdehyde; MMT = 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide; NC = negative control; OGD = oxygen and glucose deprivation; qRT-PCR = quantitative reverse transcription polymerase chain reaction; ROS = reactive oxygen species.
*p<0.05; †p<0.01; ‡p<0.001.

AC005332.7 impeded ferroptosis in oxygen and glucose-deprivation-induced AC16 cells via elevating cyclin D2 expression
Next, we investigated the role of CCND2 in AC005332.7 overexpression-mediated ferroptosis in AC16 cells following OGD treatment. Initially, sh-CCND2 transfection initially reduced CCND2 expression in AC16 cells (Figure 5A and B). Afterward, AC16 cells were transfected with lentiviruses carrying oe-AC005332.7 or with sh-CCND2 under OGD conditions. qRT-PCR and western blot were displayed that CCND2 silencing counteracted AC005332.7-mediated elevation of CCND2 expression in OGD-induced AC16 cells (Figure 5C and D). AC005332.7 overexpression promoted cell viability in OGD-induced AC16 cells, which was compromised by CCND2 knockdown (Figure 5E). CCND2 knockdown abolished AC005332.7 overexpression-mediated decrease of LDH in OGD-induced AC16 cells (Figure 5F). Moreover, overexpression of AC005332.7-mediated inhibitory effects of ROS, MDA, iron, and ACSL4 and promoting effects of GSH and GPX4 were restored by CCND2 knockdown (Figure 5G-K). We summarized that AC005332.7 suppressed ferroptosis in OGD-induced AC16 cells via elevating CCND2 expression.
Figure 5
AC005332.7 impeded ferroptosis in OGD-induced AC16 via elevating CCND2 expression. (A, B) qRT-PCR and western blot were conducted to detect CCND2 expression. (C, D) qRT-PCR and western blot were used for detecting CCND2 expression. (E) MTT was conducted to evaluate cell viability. (F-J) The LDH, ROS, GSH, MDA, and iron levels were investigated using corresponding kits and methods. (K) Western blot was applied to examine GPX4 and ACSL4 expression. Data were shown as mean±standard deviation (n=3).
ACSL4 = acyl-CoA synthetase long chain family member 4; CCND2 = cyclin D2; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; GPX4 = glutathione peroxidase 4; GSH = glutathione; LDH = lactate dehydrogenase; MDA = malondialdehyde; MTT = 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide; NC = negative control; OGD = oxygen and glucose deprivation; qRT-PCR = quantitative reverse transcription polymerase chain reaction; ROS = reactive oxygen species.
*p<0.05; †p<0.01; ‡p<0.001.

AC005332.7 overexpression restrained ferroptosis in an animal model of acute myocardial infarction
Mice were subjected to LAD ligation to establish an AMI model, and then infarcted myocardium was injected with lentivirus-packed oe-AC005332.7. After three days, the myocardial samples were collected for subsequent experiments. The results revealed that AC005332.7 expression was significantly down-regulated in the AMI group, whereas oe-AC005332.7 transfection abolished this downtrend (Figure 6A). In the meantime, CCND2 expression obviously declined in the AMI group compared to that in the Control group, but the overexpression of AC005332.7 reversed the expression of CCND2 (Figure 6B). H&E staining exhibited LAD ligation resulting in conspicuously disordered myocardial structure, while AC005332.7 upregulation observably improved this injury for myocardial structure (Figure 6C).
Figure 6
AC005332.7 overexpression restrained ferroptosis in an animal model of AMI. (A) qRT-PCR was performed to detect AC005332.7 expression. (B) Western blot was used for measuring CCND2 expression. (C) H&E staining was conducted to evaluate myocardial damage. (D) Immunohistochemical staining examined the expression of Ki67, CCND2, and GPX4. (E-J) LDH activity and CK, ROS, GSH, MDA, and iron levels were evaluated using corresponding kits and methods. (K) Western blot was applied to examine GPX4 and ACSL4 expression. Data were shown as mean ± standard deviation (n=9).
ACSL4 = acyl-CoA synthetase long-chain family member 4; AMI = acute myocardial infarction; CCND2 = cyclin D2; CK = creatine kinase; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; GPX4 = glutathione peroxidase 4; GSH = glutathione; H&E = hematoxylin and eosin; LDH = lactate dehydrogenase; MDA = malondialdehyde; MTT = 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide; NC = negative control; OGD = oxygen and glucose deprivation; qRT-PCR = quantitative reverse transcription polymerase chain reaction; ROS = reactive oxygen species.
*p<0.05; †p<0.01; ‡p<0.001.
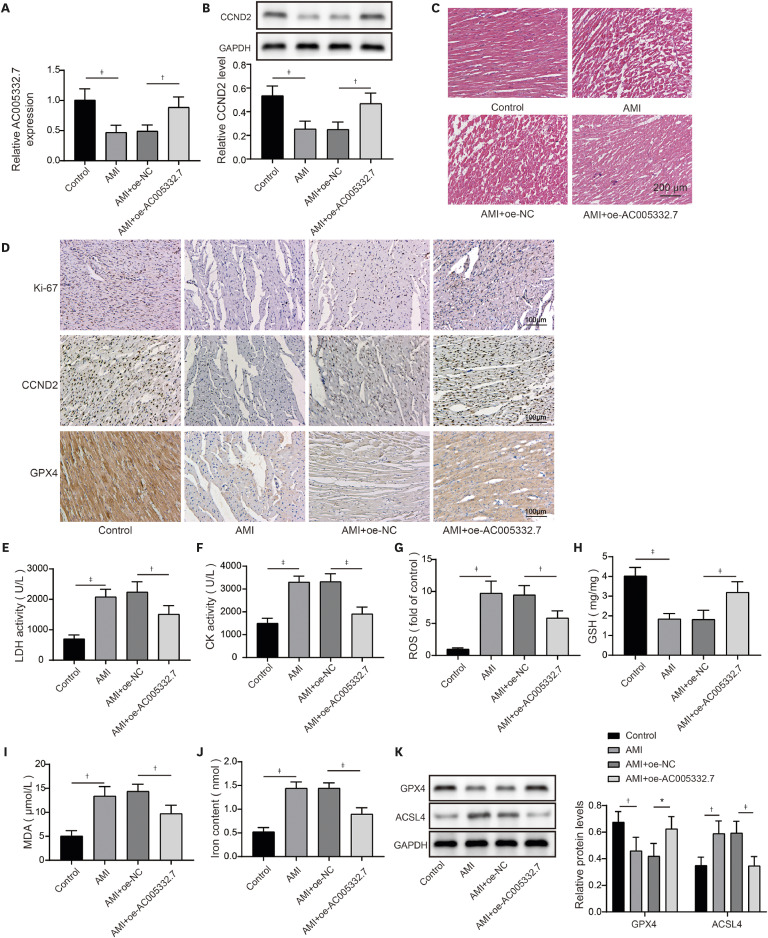
Moreover, we detected the expression of Ki67, CCND2, and GPX4 using immunohistochemical staining. IHC results exhibited that Ki67, CCND2, and GPX4 expression were decreased in the AMI group, which were reversed after overexpressing AC005332.7 (Figure 6D). Of note, we observed that LDH activity and CK production in serum were boosted in the AMI group, while these impacts were abolished by AC005332.7 overexpression (Figure 6E and F). LAD ligation resulted in the elevation of ROS, MDA, iron, and reduction of GSH in myocardial tissue. However, these changes were all compromised by AC005332.7 overexpression (Figure 6G-J). Furthermore, the western blot revealed that the GPX4 level was decreased while the ACSL4 level was increased in the AMI group, while these alterations were abolished by AC005332.7 overexpression (Figure 6K). Altogether, AC005332.7 overexpression alleviated the injury of AMI by suppressing ferroptosis.
DISCUSSION
AMI contributes to many cardiomyocyte necroses, leading to cardiac dysfunction and serious consequences for patients.16) Taking adequate measures to inhibit cardiomyocyte apoptosis and improve cardiomyocyte viability could improve AMI's prognosis. At present, ferroptosis has been reported to play an indispensable role in the development of AMI, establishing a positive correlation between ferroptosis and AMI.17) However, the detailed mechanism of ferroptosis in AMI needs further investigation. Our findings revealed that AC005332.7 regulated miR-331-3p/CCND2 axis to promote cell viability and suppress ferroptosis, thereby alleviating AMI.
LncRNAs have been extensively probed in AMI. For example, lncRNA ANRIL could largely inhibit cell apoptosis caused by AMI by modulating IL-33/ST2.18) Knockdown of MALAT1 also reduces cell apoptosis via sponging miR-320 and alleviate AMI.19) In this study, we observed that AC005332.7 expression declined in AMI cell and mouse models. Additionally, massive evidence has determined that lncRNAs could mediate ferroptosis to regulate multi-diseases, including cancer, sepsis-associated encephalopathy, and cardiac I/R injury.20)21)22) Zhang et al.22) revealed that lncRNA Mir9-3hg carried by BMSC-derived exosomal alleviated cardiac I/R injury via repressing cardiomyocyte ferroptosis. In this work, we found that ferroptosis was significantly triggered in AMI cell and mouse models. Moreover, AC005332.7 overexpression restrained ferroptosis in OGD-induced AC16 cells and LAD ligation-operated mice and improved myocardial damage, suggesting AC005332.7 functioned as a protective molecule in AMI. Our results firstly explored the functions of AC005332.7 and the regulatory relationship between AC005332.7 and ferroptosis in AMI.
In recent decades, accumulating evidence has determined that the mechanism of competing for endogenous RNAs (ceRNAs) is extensively involved in regulating multi-diseases such as myocardial infarction, acute lung injury, and cancer.23)24)25) CeRNAs mainly refer to that lncRNAs or circular RNAs can competitively bind to miRNA and interfere with miRNA binding to mRNA, thereby regulating genes’ expression and further affecting cell functions.26) For example, lncRNA MIAT acts as a ceRNA to mediate Wnt5a expression by sponging miR-488-3p in hypoxia-induced cardiomyocyte injury.5) Here, miR-331-3p was determined as the downstream molecule of AC005332.7 by dual luciferase reporter gene assay and RIP assay. Moreover, AC005332.7 could negatively regulate miR-331-3p expression in AC16 cells. Until now, the investigations of miR-331-3p mainly focused on cancer, mediating multi-functions such as cell proliferation and apoptosis.27) A previous study described an increase of miR-331-3p in ST-segment elevation myocardial infarction of patients,7) indicating miR-331-3p might be involved in AMI. Therefore, in this study, we investigated the role of miR-331-3p in AMI. Our results exhibited abnormal elevation of miR-331-3p in AMI. Furthermore, miR-331-3p overexpression reversed AC005332.7-mediated promotion of cell viability and repression of ferroptosis in OGD-induced AC16 cells, which firstly built up a linkage between AC005332.7 and miR-331-3p as well as mediating ferroptosis in AMI.
CCND2 is a crucial molecule in AMI. A study figured out that overexpression of CCND2 mitigated the myocardial injury caused by myocardial infarction.28) In our study, CCND2 expression was presented reduction in AMI in vitro and in vivo. As previously depicted, CCND2 mediated by miRNA let-7i-5p promote cardiomyocyte proliferation and suppress cardiomyocyte injury in multiple angio-cardiopathy.29) In our study, we found miR-331-3p could directly target CCND2 and negatively modulate its level in AC16 cells. Of note, Ni et al.12) proposed that lncRNA ZFAS1 indirectly upregulated CCND2 expression by suppressing miR-150-5p to resist diabetic cardiomyopathy via inactivating ferroptosis. Here, CCND2 silencing abolished AC005332.7-mediated suppressive ferroptosis in cardiomyocytes under OGD condition.
In conclusion, we first put forward that AC005332.7/miR-331-3p/CCND2 axis was engaged in inhibiting ferroptosis in AMI, which could largely reduce cardiomyocyte injury in vitro and in vivo, which might provide new strategies for the treatment of AMI.
 XML Download
XML Download