

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Neuromyelitis optica spectrum disorders (NMOSD, previously known as neuromyelitis optica or Devic's syndrome) are rare autoimmune diseases involving the optic nerve, spinal cord, and central nervous system (CNS) [123]. Autoantibodies against aquaporin-4 immunoglobulin G (AQP4-IgG) are important in the pathogenesis of NMOSD. The diagnostic criteria were based on the typical clinical, immunological, and radiological features [2]. Brain MRI of NMOSD patients shows typically nonspecific white matter alterations, such as patches and dots, in the subcortical and deep white matter [4]. Brain lesions are often observed as multiple enhanced mass; therefore, differentiating from multiple brain tumors, such as brain metastases, lymphomas, and high-grade gliomas, is necessary. The accurate diagnosis of NMOSD through detailed medical history, laboratory test, and radiologic findings may reduce the rates of false diagnosis and unnecessary test or treatment.
We report a rare case of NMOSD, a disease of antibody-mediated disorder, mimicking multiple brain tumors.
Go to : 
CASE REPORT
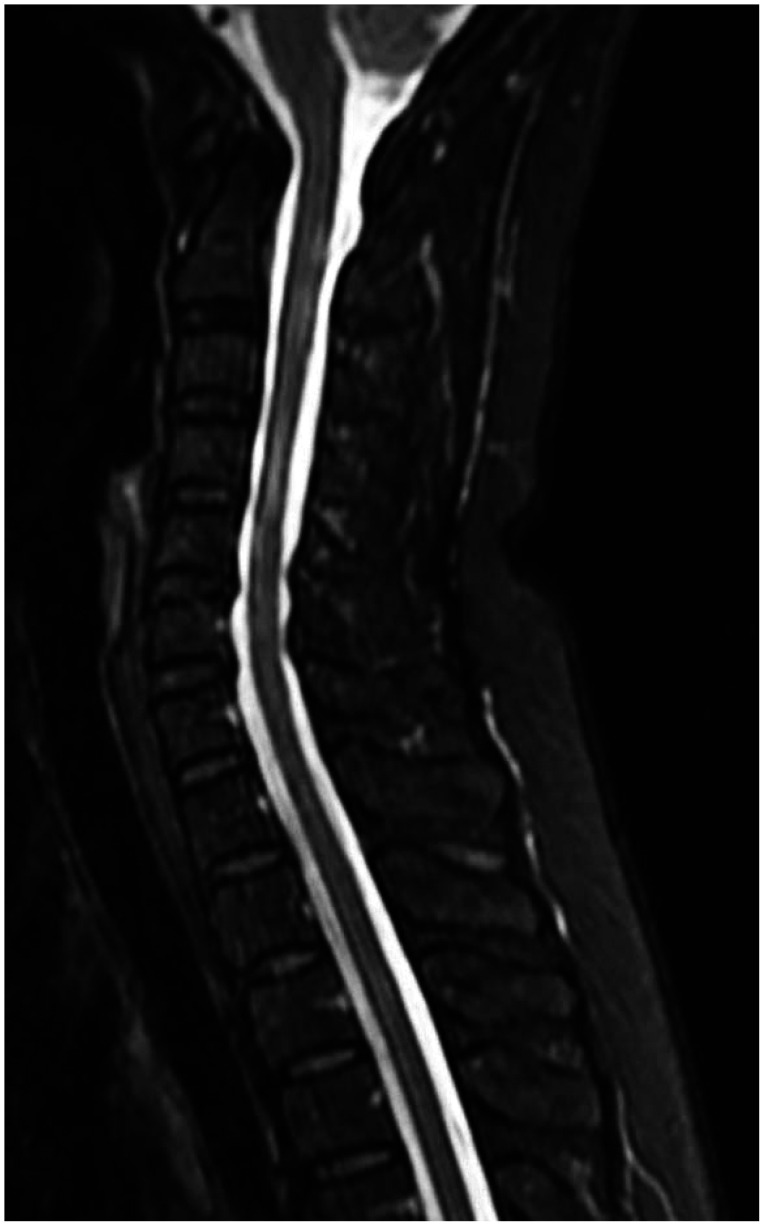
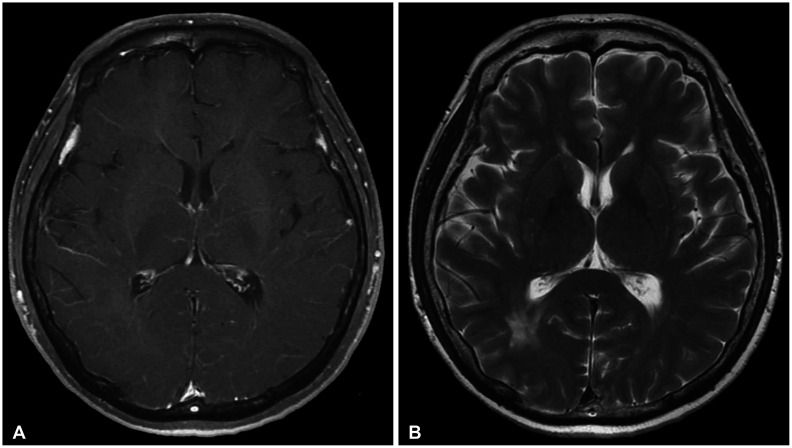
A 53-year-old woman, who was diagnosed with multiple brain tumors in a local clinic, was referred to our hospital for further diagnosis and treatment. She had originally visited the clinic for weakness and paresthesia in her right arm and leg and had been initially diagnosed with multiple brain metastases. She experienced a decreased vision in her left eye a few years ago. Her brain MRI showed multiple enhancing masses in the right parahippocampal gyrus, occipitotemporal area, and left frontal lobe, which were considered multiple brain metastases under consultation with a radiologist (Fig. 1). In addition, small T2 high signal lesions in the subcortical white matter also showed subtle enhancement, which were considered small metastatic lesions. Subsequently, chest-abdomenpelvis CT revealed no evidence of primary tumor of metastases. The cervical spine MRI taken to examine her right paresthesia shows multiple abnormal signal changes in the spinal cord over C2–T7, which was similar to acute myelitis (Fig. 2). In her medical history, a decreased vision was observed in her left eye from several years ago. Upon ophthalmologic examination, she was diagnosed with optic neuritis. Laboratory examination showed normal findings including negative AQP4-IgG. With clinical characteristics involving the optic nerve, spinal cord, and brain, and typical radiological appearances of the lesions, the multiple brain lesions were diagnosed as NMOSD. Intravenous corticosteroid was administered to treat an acute attack. As maintenance treatment, oral steroid and immunosuppressive drug (azathioprine) have been administered. A 2-year follow-up brain MRI reveals the disappearance of the previously enhanced lesions, but T2 high signal lesions are still observed (Fig. 3).
 | Fig. 1Brain MR images (A-C) show multiple enhancing masses in the right parahippocampal gyrus, occipitotemporal area, and left frontal lobe, which are considered multiple brain tumors. Nonspecific small dots and patches in the subcortical and white matter (D) are also seen.
|
The Institutional Review Board of Kyungpook National University Hospital exempted informed consent due to its retrospective nature and minimal risk for harm to the patient, and this report was conducted according to the guidelines of the Declaration of Helsinki for biomedical research.
Go to :
DISCUSSION
NMOSD is a rare autoimmune disorder involving the optic nerve, spinal cord, and CNS [123]. In the past, it was considered as a form of multiple sclerosis (MS). Now, NMOSD can be distinguished from MS, based on clinical, radiological, and immunological findings. AQP4-IgG test is an important diagnostic tool, but in the novel classification, both groups (with and without AQP4-IgG) are eligible for NMOSD diagnosis [12]. The first international consensus criteria published in 2015 based the diagnosis of NMOSD on the presence of core clinical characteristics, AQP4 antibody status, and MRI features (Table 1) [2]. The diagnostic criteria for NMOSD with negative or unknown AQP4-IgG status are more exacting. This patient was diagnosed through the diagnostic criteria for NMOSD with negative or unknown AQP4-IgG status.

Table 1
NMOSD diagnostic criteria for adult patients
AQP4, aquaporin-4; IgG, immunoglobulin G; LETM, longitudinally extensive transverse myelitis lesions; NMOSD, neuromyelitis optica spectrum disorders. Adapted from Wingerchuk et al. Neurology 2015;85:177-89 [2].
![]()
Neurological symptoms, such as encephalopathy and brainstem alteration, develop in 15% of NMOSD patients [5]. Brain lesions are observed in the brain MRI of about 89% of AQP4-negative NMOSD patients [6]. In these patients, the accurate diagnosis through brain MRI findings is very important to differentiate between AQP4-negative NMOSD and other autoimmune diseases and MS [5]. The most common findings in NMOSD were nonspecific small dots and patches in the subcortical and deep white matter. In 9–36% of NMOSD patients, contrasts-enhancing brain lesions are reported [6]. Differentiating these brain lesions from multiple brain tumors is important, as in this case. Characteristic brain MRI findings in NMOSD include periependymal lesions surrounding the ventricles, hemispheric tumefactive lesions, involvement of corticospinal tracts, and cloud-like enhancement [4]. A poorly marginated, subtle, and multiple patchy pattern was called cloud-like enhancement. Brain lesions are generally located close to the ventricles, in the diencephalon and hypothalamus, which are not characterized by central veins. Moreover, cortical lesions are absent in NMOSD [7].
The spine MRI shows characteristic longitudinally extensive lesions (more than three vertebral segments) and central/gray involvement in the spinal cord of NMOSD patients. Sometimes, continuous lesions in the spinal cord appear as contrast-enhanced lesions in NMOSD patients, which are often misdiagnosed as a malignant spinal cord tumor; thus, a surgical biopsy is performed. Of course, it occasionally occurs in the brain.
Although optic nerve involvement is an important clinical finding for the diagnosis of NMOSD, diagnosing optic neuritis in brain MRI is difficult. Optic neuritis in NMOSD patients may show normal brain MRI, as in this case [8]. In patients with multiple brain lesions mimicking brain tumors, checking whether ophthalmic symptoms exist is important, even if brain MRI findings show a normal optic nerve. In addition, if abnormal findings on the spine MRI taken after checking the medical history of the spine are found, the NMOSD diagnosis becomes more conclusive.
Discussing the typical radiological findings of NMOSD is of great help in differentiating NMOSD from other diseases, which can reduce unnecessary examinations, such as surgical biopsies. Multiple brain lesions in both hemispheres are likely to be mistaken for malignant brain tumors. To differentiate NMOSD from multiple brain tumors, brain and spine MRI should always be performed. In our study, the entire CNS MRI (brain and spine) was very important to help distinguish them from other diseases. Metastatic brain tumors were a first impression even prior to the spine MRI examination.
As was the case in our patient, NMOSD does not require surgical treatment. The accurate diagnosis of NMOSD was achieved through characteristic brain and spine MRI, ophthalmic examination, and medical history without pathological examination. The precise review of brain and spine MR images in particular can lead to the correct diagnosis of NMOSD.
In conclusion, we report a rare case of NMOSD mimicking multiple brain tumors. NMOSD should be considered in the differential diagnosis of multiple brain lesions in both hemispheres. Sufficient medical history and spine as well as brain MRI are essential in differentiating NMOSD from multiple brain tumors.
Go to :
 XML Download
XML Download