

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Dysembryoplastic neuroepithelial tumors (DNETs) are a distinct type of low-grade glioma in terms of histology and clinical behavior. It belongs to the glioneuronal and neuronal tumor category in World Health Organization (WHO) classification of brain tumors. Slow-growing brain tumors arising from the cerebral cortex, associated with chronic epilepsy in children have been described since the 19th century [1]. Many of the tumors are glioneuronal tumors such as gangliocytoma and ganglioglioma. In 1988, Daumas-Duport et al. [2] reported the presence of a particular type of glioneuronal tumor, distinguished from more common gangliogliomas and called the new entity a DNET. More than 30 years have passed since the publication of the original article, but a DNET, as a disease entity, has survived the test of time, during which 5 revised versions of the WHO classification of brain tumors have emerged [34]. In this review, we will introduce the general features of DNET and discuss some new findings in molecular biology and clinical practice on DNET.
Go to : 
CLINICAL FEATURES
DNET is a relatively uncommon type of brain tumor. The original study of Daumas-Duport et al. [2] comprised 39 patients from the epilepsy surgery registry of St. Anne Hospital and the brain tumor registry of Mayo Clinic; both were collected for decades. In fact, the patient numbers in many studies on DNET do not exceed a few dozen. In the Korean brain tumor registry, mixed glioneuronal tumors accounted for 7.0% of all gliomas diagnosed in 2013, but it was not specified how many DNETs were included [5]. Focusing on childhood tumors, DNET accounted for 1.5% of all brain tumors and approximately 30% of mixed glioneuronal tumors in the pediatric brain tumor registry of Seoul National University Children’s Hospital [6].
DNET develops predominantly in children and adolescents. Young adults are occasionally diagnosed with DNETs, but most patients develop seizure attacks earlier than age 20. A strong association with chronic epilepsy is one of the defining features of DNET. Daumas-Duport et al. [2] emphasized that all of their patients presented with complex partial seizures except for 2 patients with headaches. In 2003, Luyken et al. [7] reported a large spectrum of brain tumors associated with chronic epilepsy. They coined the term long-term epilepsy-associated tumors (LEATs) to designate DNET, ganglioglioma, pleomorphic xanthoastrocytoma, and other rare glioneuronal tumors frequently found in patients with drug-resistant epilepsy. Although the concept of LEAT has been questioned primarily because of the heterogeneity of this clinical group, DNET is the representative tumor of LEATs with its strong epileptogenicity and benign nature [8]. DNETs occupied 5.9% of the European Epilepsy Brain Bank tissue registry, an extensive tissue cohort obtained from epilepsy surgery, and rank the fourth in the causes of epilepsy, next to hippocampal sclerosis, ganglioglioma, and focal cortical dysplasia (FCD) [9].
DNETs develop in the cerebral cortex, involving a whole gyrus or a wedge-shaped part of a cerebral lobe. The temporal lobe is the most common location, although any part of cerebral cortex can be involved [10]. Cortical location and predilection to the temporal lobe make patients with DNETs highly vulnerable to epileptic seizures. Another reason for its high epileptic potential is the dysplastic nature of DNET [2]. Typical DNETs comprise the tumor core surrounded by the rim of dysplastic cortex corresponding to FCD (a complex-type DNET) [11]. The dysplastic cortex around DNETs is regarded as highly epileptogenic [12]. Some DNETs lack a surrounding dysplastic cortex (a simple-type DNET) and epileptogenic zone is co-localized with the tumor extent [13].
Focal seizures with or without impaired awareness are the most common symptoms of DNET. Secondary generalization is infrequent. Patients with DNETs usually progress into chronic drug-resistant epilepsy [11]. Headache is a rare complaint only for very large tumors. Focal neurologic deficits such as motor weakness are seldom observed in tumors near the primary motor cortex.
Go to :
IMAGING FINDINGS AND SATELLITE LESIONS
CT shows a mass lesion of low attenuation involving cortex and subcortical area. MRI is the mainstay of diagnostic modality for DNET. DNETs typically have a bubble-like, or popcorn-like multinodular shape with low signal intensity in T1-weighted images and high signal intensity in T2-weighted and fluid-attenuated inversion recovery (FLAIR) images (Fig. 1). DNETs are poorly enhanced with gadolinium, but some tumors show patchy or partial enhancement. Gangliogliomas also involve cerebral cortex and subcortical white matter with a predilection to the temporal lobe. A small cystic or nodular architecture is characteristic for gangliogliomas, in contrast to the multicystic structure of DNETs. Strong gadolinium enhancement is more common (approximately 50%) in gangliogliomas [14]. However, it is difficult to distinguish non-enhancing gangliogliomas from DNETs and even from the FCD [14]. 11C-methionine positron emission tomography (PET) is helpful because gangliogliomas show hypermetabolism whereas DNETs exhibit variably low metabolism [15].
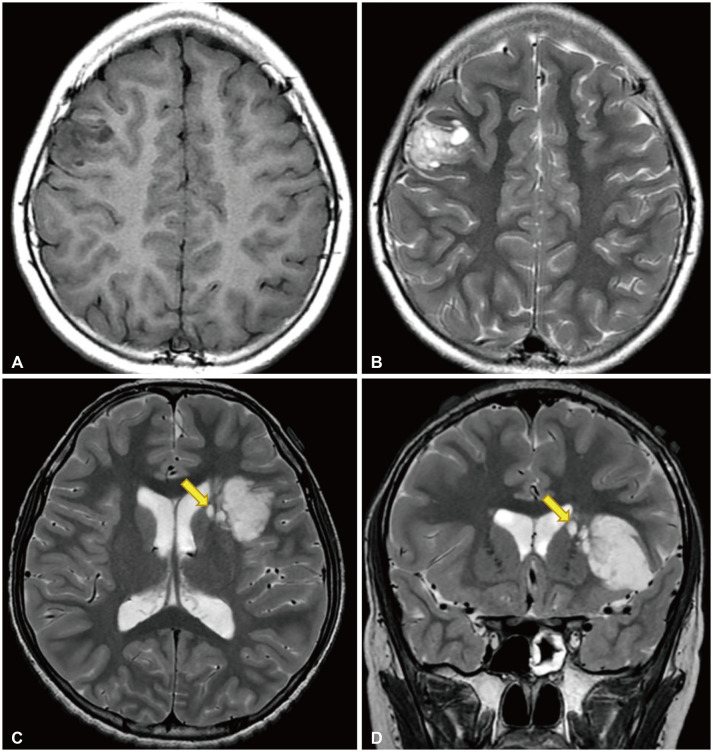 | Fig. 1Radiological features of dysembryoplastic neuroepithelial tumor. A and B: T1-weighted and T2-weighted magnetic resonance images of a 10-year-old girl who presented with recurrent focal seizures. A multinodular mass is located in the right middle frontal gyrus. C and D: T2-weighted axial and coronal images of a 12-year-old boy who came to the clinic for chronic epilepsy. A large tumor is situated in the insular and left inferior frontal lobes. There are several satellite lesions (yellow arrows) on the medial side of the mass, in the caudate nucleus and internal capsule.
|
There are many variants in the shape of DNETs. Chassoux et al. [16] classified the MRI morphology of DNETs into 3 categories: Type 1 polycystic, Type 2 nodular, and Type 3 diffuse (dysplastic). They correlated Type 1 with simple and complex histology and Type 2 and 3 with nonspecific histology, and the MRI types were highly associated with the extent of epileptogenic zone and surgical outcomes in terms of seizure control. However, the classification has not been widely used and the clinical correlation remains to be verified.
In the original study, Daumas-Duport et al. [2] reported the presence of nodular foci beneath the DNET mass. They showed that the nodular foci consisted of multiple cell types, i.e., neurons, astrocytes, and oligodendrocytes. Sometimes, gliomalike histology was observed in the nodular foci. Later, Urbach et al. [14] emphasized the multinodular architecture of DNET on MRI. They pointed out that sometimes single smaller cysts are observed in the vicinity of the tumor, from which they are clearly separated. We found that such small nodular foci are found in up to 50% of DNETs on high resolution MRI [17]. We called the foci ‘satellite lesions’ because they are clearly separated from the tumor core by a thin white matter, documented on 3-planar MRI and intraoperative findings (Fig. 1). Removal of the satellite lesions may injure important functional tracts in the deep subcortical area and residual satellite lesions significantly impact tumor control and seizure control [17].
Go to :
PATHOLOGICAL FEATURES
According to the WHO classification of brain tumors, DNET was described as below: the histopathological hallmarks are the multinodular intracortical growth pattern (Fig. 2) and the pathognomonic specific glioneuronal element, i.e., the presence of columns oriented perpendicularly to the cortical surface, formed by bundles of axons lined by small oligodendroglia-like cells (OLCs) [18]. Between these columns, neurons with normal cytology appear to float in a mucoid matrix (floating neurons) (Fig. 3). Atypical features such as mitoses, high cellularity, necrosis, or eosinophilic granular bodies are absent or rare. To summarize, DNET has the following three characteristic findings: 1) multinodular intracortical pattern, 2) glioneuronal columns with OLCs, and 3) floating neurons in the mucoid matrix (mucin matrix) (Fig. 4).
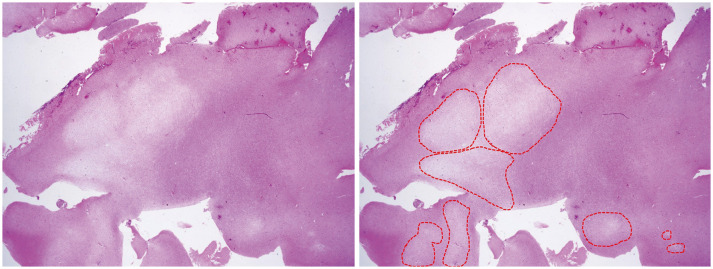 | Fig. 2Scan view of the dysembryoplastic neuroepithelial tumor (H&E staining; ×12.5). The right side shows multinodular intracortical growth pattern (encircled by red dots).
|
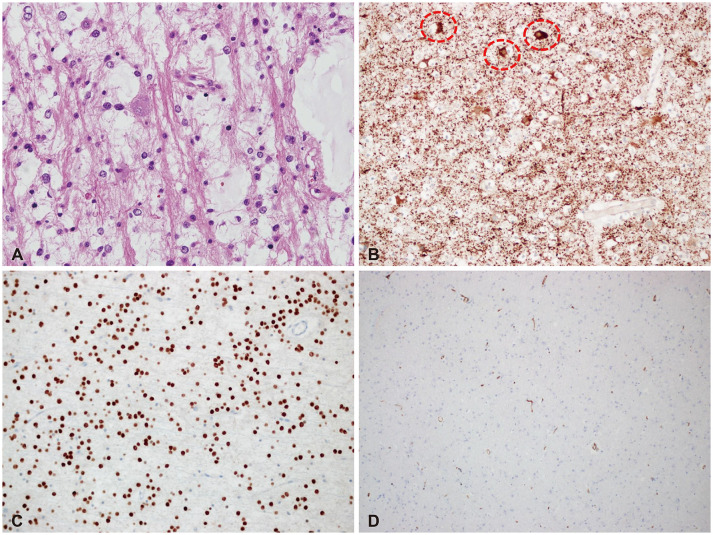 | Fig. 3Typical histological and immunohistochemical findings of dysembryoplastic neuroepithelial tumor. A: A high-power view (H&E staining; ×400) shows glioneuronal elements (columns formed by bundles of axons lined by oligodendroglia-like cells [OLCs] and floating neurons in the multinodular growth pattern). B: Faint MAP2 expression in OLCs and marked expression in floating neurons (encircled by red dots) are noted (MAP2; ×200). C: OLIG2 is expressed in the OLCs (OLIG2; ×200). D: CD34 is negative in some cases. However, it can show variable expression (CD34; ×200).
|

DNET has several histological forms including simple, complex and diffuse/non-specific. Unlike the simple form with the typical histological features described above, the remaining forms have other histological attributes or lack the typical glioneuronal element. There are many controversies between even experienced neuropathologists about the validity of the subtypes of DNET. In addition, DNETs share a lot of similar histological features with other tumors, especially gangliogliomas. These points are one of reasons for the introduction and upgrade of the term LEAT to ‘low-grade, developmental, epilepsy-associated’ brain tumors [8].
OLCs in a DNET are neither mature glial cells nor neuronal cells. Therefore, OLCs show glial markers OLIG2 and PDGFRA but not GFAP. MAP2 expression is typically faint in OLCs. The floating neurons can express NeuN. DNETs show variable BRAF (V600E) and CD34 immunoreactivities which are more commonly expressed in gangliogliomas [18]. If there is expression of IDH (R132H), it is not DNET.
With new technologies and molecular knowledge, the above-mentioned term LEAT can be more subdivided and classified [18]. DNETs have FGFR1 alterations including internal tandem duplication, fusion, and missense mutation [1920]. The tyrosine kinase domain of FGFR1 is particularly known to be specific to DNET, although not exclusively. Recently, DNA methylation or transcriptional profiling has shown a distinct pattern of DNET, and they can be helpful for the diagnosis of unresolved lesions. The new 2021 WHO classification of brain tumors suggests the diagnostic criteria (Fig. 5) [18].
 | Fig. 5Diagnostic criteria for DNET (according to the 2021 WHO classification). DNET, dysembryoplastic neuroepithelial tumor. Adapted from Piertsch et al. Central Nervous System Tumors (5th ed); 2021. p. 123-6, with permission of International Agency for Research on Cancer [18].
|
Go to :
TREATMENT AND OUTCOME
DNET is a benign, low-grade glioma with an excellent prognosis. Although oncogenic mutations/duplication of the FGFR1 gene is documented in the majority of DNETs, the tumor is still regarded as a dysplastic neoplasm with limited, very low malignant potential. Some reports of the malignant transformation of DNET are anecdotal and the validity of the initial diagnosis should also be reconsidered [2122]. Daumas-Duport et al. [2] emphasized the benign nature of the disease and reported that 44% of their patients had an incomplete resection but no clinical or radiological progression was observed. The clinical stability of DNET has long been regarded as true. However, evidence has accumulated that tumor progression is common after incomplete resection, and reoperations for seizure recurrence are also not uncommon [1723]. A small but meaningful series from St. Jude Children’s Research Hospital showed that tumor recurrence/progression occurred in 4 (36%) out of 11 patients [24].
Many studies have emphasized that complete resection is crucial factor for tumor control and seizure outcome. Gross total resection was reported in 60%–90% of patients with DNETs [25]. However, complete resection of DNETs is often hampered by ill-defined borders in T2/FLAIR images, multi-gyral involvement, tumor location in the eloquent cortex, and the presence of satellite lesions. As mentioned above, satellite lesions are mainly located on the medial side of the tumor, sometimes near the ventricle, where the pyramidal tract or optic radiation can be encountered [17]. Some remnant satellite lesions did not grow during the follow-up, but we experienced slow but steady growth of a residual satellite lesion that led to overt tumor recurrence and reoperation.
After surgery, the reported seizure freedom rates were 60%–80% [25]. Even partial removal of the tumor can give sufficient seizure control to the patients, although temporarily. However, tumor progression is often heralded by a breakthrough seizure attack with or without antiseizure medications. Therefore, complete resection is of utmost importance for long-term seizure freedom [17]. The extent of resection beyond the DNET margin to achieve a good seizure outcome has been controversial. A wider cortisectomy beyond the tumor boundary is required for DNETs for adequate seizure control because DNETs are accompanied by a surrounding rim of the dysplastic cortex where the ictal onset zone is located [26]. Tailored resection is recommended based on invasive monitoring or electrocorticography data to delineate the ictal onset zone [27]. However, lesion (tumor)-directed surgery can also be applied to many patients because complete tumor resection yields favorable seizure control. The presence of satellite lesions is a poor prognostic factor for seizure control after surgery [17]. Whether this reflects the difficulty of complete resection and subsequent seizure recurrence, or the presence of widespread pathologic lesions beyond the observable satellite lesions is unclear but is an interesting question.
Go to :
CONCLUSIONS
The distinctive clinical features and histological architectures make DNET a unique and important entity among a variety of brain tumors. DNETs are highly associated with chronic epilepsy, and molecular research revealed that DNETs are driven by one or a few oncogenic mutations. Recent studies reported higher recurrence rates of DNETs than previously thought. It is true that complete resection leads to long-term tumor control and seizure freedom, but there are still questions on how to achieve total resection for DNETs with indistinctive borders, with satellite lesions, or located in the eloquent cortex. Piecing together the histology, molecular biology and imaging studies will produce meaningful answers to the unsolved queries.
Go to :
 XML Download
XML Download