

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Brain metastases (BMs) carried a dismal prognosis in the 20th century when the tumors could be treated palliatively by decompressive surgery with or without conventional whole-brain radiation. The introduction of stereotactic radiosurgery into clinical practice in the 1990s improved local control of BM, resulting in prolonged overall survival of patients with BM [12].
However, as chemotherapeutic agents can hardly penetrate the blood–brain barrier (BBB), the brain became a sanctuary for metastasis in long-term cancer survivors as well as an immune refuge due to limited exposure to circulating antigens and immune cells, protecting BM and allowing it to grow [3]. For certain cancers with mutations, for which there are targeted therapies, the outcomes of patients with BM have markedly improved over the last decade due to these immune and targeted therapies. However, even in the era of targeted therapy, BM is frequent in HER-2+ breast cancer and advanced epidermal growth factor receptor–mutated or anaplastic lymphoma kinase–rearranged non-small-cell lung cancers (NSCLCs) [45]. Research has suggested that a discrepancy between BM tissue and primary tumors contributes these heterogeneous therapeutic responses. Furthermore, despite successful initial treatment for BM, recurrent BM, either local or distant, can hardly be treated completely [67].
Thus, the biological and molecular mechanisms underlying the metastasis of cancer cells to the brain are essential to the development of target therapy and/or preventing recurrence, though they remain unclear. An accurate genetic/epigenetic analysis of BM tissue compared with matched primary cancer, if possible, has become valuable, and understanding how BM cancer cells adapt to the inhospitable brain tumor microenvironment (TME) and modulate immune surveillance is crucial.
BRAIN ORGANOTROPISM OF METASTATIC CANCER CELLS
Disseminated tumor cells (DTCs), which are physically separated from the primary tumor mass, spread to other anatomical locations through a hematogenous route (blood circulation). It is well known that tumors of different origins display unique patterns of metastasis with a preference for a particular set of organs, such as colon cancer to the liver and prostate cancer to the bones [8]. This organotropism of DTCs can be partly explained by the anatomical proximity of organs but mainly by the “seed and soil” theory, which necessitates a proper TME for cancer cells to survive and grow. The hematogenous route is the most frequent way for DTCs to reach the brain. BMs tend to distribute along gray-white junctions and watershed vascular distributions where DTCs lodge in capillary beds [9]. The distribution of BMs follows cerebral blood flow and the absence of a lymphatic system in the brain supports this hematogenous route. If metastasis occurred only by chance, the liver, which has fenestrated sinusoidal endothelial and receives a major venous influx, would have metastasis equal to or more than that of the lungs. Furthermore, the brain, protected from materials in the circulating blood by the BBB, should have little chance of metastasis. However, the obvious difference of proportional BM incidence according to the type of primary cancer, such as NSCLC, breast cancer, and melanoma, denies this simple “by chance” hematogenous spread but supports “seed and soil” tropism. In a mouse BM model, cell lines among the same primary origin showed organotropism: e.g., MDA-MB-361 for the brain and MDA-MB-231 for the bones and lungs among breast cancer cell lines, and a cell line derived from BM revealed a high propensity for establishing BM compared with other cell lines [410].
Cancer cells of common origin show different gene expression patterns upon metastasis to different organs as a result of interaction between cancer cells and the TME. Basnet et al. [11] verified that early micrometastases in the brain and lungs derived from intracardiacally injected MDA-MB-231 cells showed a remarkable difference in gene expression patterns using the in-situ Flura-seq technique. Thus, rather than tail vein injections or an orthotropic model, carotid artery injections are the preferred route to establish a BM animal model, study metastatic processes, or evaluate the therapeutic effects of drugs in preclinical development. Zhang et al. [12] verified that tumor cells with intact phosphatase and tensin homolog (PTEN) lose that expression after dissemination to the brain via internal carotid artery injection but not to other organs. Furthermore, the process was mediated by exosome-contained microRNA secreted by astrocytes. This brain TME-induced PTEN mRNA and protein down-regulation were reversible after the tumor cells left the brain. A similar observation was reported by Jin et al. [13], who verified that p-Akt expression in primary NSCLC tissue differs among patients and correlates with the risk of BM: patients exhibiting medium to high p-Akt expression had a higher incidence of BM than those with low to no p-Akt expression.
Brain tropism of DTCs is also influenced by the primary TME. Sevenich et al. [14] reported that high cathepsin S expression at the primary site correlated with decreased BM-free survival in breast cancer patients. Cathepsin S, which was differentially expressed by repeated implantation of organ-specific tumor cells using a protease mRNA microarray, promoted the transmigration of DTCs through the BBB by inducing cleavage of intercellular tight junctions. In their mouse model, only the combined depletion of both macrophages and tumor cathepsin S significantly reduced BM.
GENETIC EVOLUTION AND HETEROGENEITY OF BM
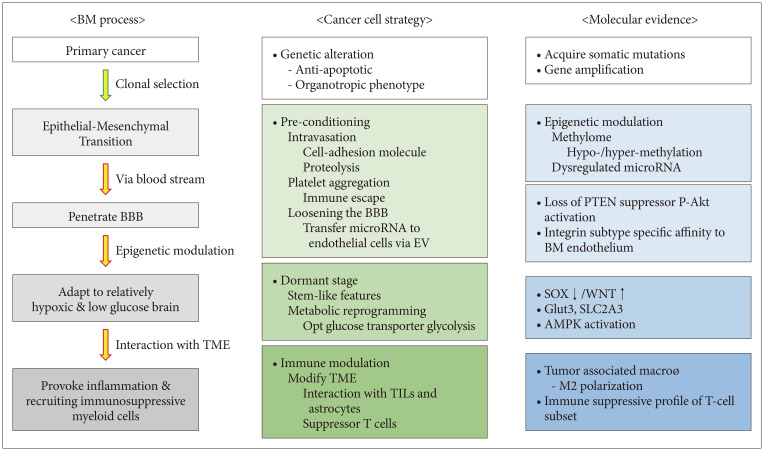
It is well known that cancer develops through a process of somatic evolution [15]. Early oncogenesis is characterized by mutations in a constrained set of driver genes and specific copy number gains. In cancer evolution, including metastasis, the mutational spectrum changes significantly and diversely as the tumor adapts to the TME, forced by genomic instability. Robinson et al. [16] performed comprehensive integrative sequencing (whole-exome and transcriptome sequencing) of 500 cancer patients harboring metastatic cancers. The most prevalent genes somatically altered in metastatic cancer included TP53, CDKN2A, PTEN, PIK3CA, and RB1. Putative pathogenic germline variants were present in 12.2% of cases, of which 75% were related to defects in DNA repair. The authors suggested that these genetic variations stem from the complex molecular landscape and microenvironment of metastatic cancers.
The clonal selection hypothesis suggests that metastatic cancer cells are selected toward an appropriate phenotype for cancer metastasis and chromosomal instability contributes to the development of this metastatic clone. Epithelial–mesenchymal transition (EMT) is a well-known mechanism of cancer cells to achieve distant metastasis. Metastatic clone selection and additive epigenetic modulation are the basis of this hypothesis. Brastianos et al. [17] performed comparative whole-exome sequencing of paired primary cancer and BM tissues. They verified branched evolution patterns of somatic mutation with actionable targets in approximately 2/3 of cases. Although this study was based on paired primary–BM samples to extract responsible driver mutation of BM, they failed to find a common mutation peculiar to BM compared with primary cancer likely due to the heterogeneity of primary cancer. Later, they confined primary cancer to NSCLC adenocarcinoma and performed whole-exome sequencing on 73 BM and 503 primary cancer samples [18]. They identified three regions with significantly higher amplification frequencies—MYC, YAP1, and MMP13—and significantly more frequent CDKN2A/B deletions in BM than in primary cancer. Over-expression of these three genes increased the incidence of BM in patient-derived xenograft mouse models. Jacob et al. [19] performed whole-exome sequencing in five widely used experimental metastasis models that were independently derived through in vivo selection from heterogeneous human cancer cell lines. Intracardiac injection of transplantable cancer cells from BM and exome sequencing revealed no evidence for genetic loss or gain compared with parental cells but did indicate selection for the KRASG13D mutation. They suggested that metastatic competence can stem from the selection of heterogeneous cancer cell populations without the need for the acquisition of additional mutations.
Hypoxic conditions formed in the necrotic central portion of metastatic tumors also contribute to their genetic instability. As a consequence of oxidative stress, reactive oxygen species are released during phagocytosis by macrophages in response to tumor necrosis factor. Reactive oxygen species production is suggested to increase the rate of mutation in tumor cells that can resist immune-mediated cell death [20].
EPIGENETIC MODULATION IN BM
The intercellular milieu, including endothelial cells, pericytes, fibroblasts, and leukocytes, contributes to stresses on the tumor cells, enhancing genomic instability and epigenetic dysregulation. An analysis of metastatic tumors compared with primary tumors found that acquired germline mutation is relatively rare and the methylation profiles are significantly different [16]. DNA hypomethylation in cancer often affects more of the genome than hypermethylation, so net losses of genomic 5-methylcytosine are seen in many human cancers. However, the selectivity of hypo-/hypermethylation occurrence in metastatic tumors remains unknown. Many authors are currently searching for alterations in metastatic cancer cells at the levels of mRNA expression, DNA copy number, and DNA methylation. Liu et al. [21] analyzed the methylomes of gliomas, BM, and primary cancer via relative methylation orderings of the CpG sites. Of 133 BM samples, 132 were identified as non-gliomas and 126 were correctly classified as their corresponding original cancer types. This algorithm also verified conserved methylation pattern between primary cancer and BMs. Salhia et al. [22] analyzed the methylome data of 32 breast cancer BM samples, 12 non-neoplastic brain samples, 15 non-neoplastic breast samples, and 48 early-stage primary breast cancer samples. They found that overall methylation levels were higher in the breast cancer BMs than in the other samples. Integrating DNA methylation and gene expression data revealed defects in cell migration and adhesion due to hypermethylation and down-regulation of PENK, EDN3, and ITGAM. Hypomethylation and up-regulation of KRT8 likely affect adhesion and permeability.
MicroRNAs have been highlighted for their contributions to important adaptive post-translational modifications in cancer development, metastasis, and therapeutic resistance [23]. The microRNA profile changes in metastatic tumor cells compared with those of primary tumors have been reported for many different cancer types. Many studies have shown that the brain microenvironment changes the microRNA profile of tumor cells compared with the primary tumor due to the interaction between the brain TME and the metastatic tumor cells. Although there have been tens of dysregulated microRNAs in BM, much less microRNA is differentially expressed in paired BM–primary cancer studies. Co-culture of lung cancer cells with astrocytes led to down-regulation of miRNA-768-3p, which drives KRAS expression, and miRNA-768-3p was reduced in a patient’s BM compared with normal brain tissue [24]. Zhou et al. [25] suggested that exosome-mediated transfer of cancer-secreted miR-105 efficiently destroys tight junctions and the integrity of the BBB against metastasis. Exosomes from metastatic MDA-MB-231 cells are highly enriched with miR-105 and target migration-related protein zonula occludens-1 (ZO-1, tight junction protein-1). By targeting ZO-1, cancer cells diminish tight junctions and destroy the barrier function of endothelial monolayers, leading to increased vascular permeability and promotion of metastasis.
Because a single microRNA has multiple target mRNAs, it is not easy to predict the final consequences of the summed effect of dysregulated microRNAs. MicroRNAs can aid diagnosis and monitor disease progression not only in BM tissue but also in liquid (cerebrospinal fluid) biopsy. However, in therapeutics, it is difficult to deliver mimics or antagomir intracellularly or to selectively modulate target genes.
ROLE OF PLATELETS IN EMT AND DTC LODGING IN THE CEREBRAL VASCULATURE
The tumor-platelet interaction has been linked with fibrin production and ensuing hypercoagulability in cancer patients [26]. Tumor-generated thrombin activates adhesion of circulating DTCs to platelets while both activating the platelets and stimulating tumor cell growth. Furthermore, tissue factor is upregulated in hypoxic sites, such as those within tumor tissue, which encourages platelet activation [26]. Considering the seminal role of platelets in wound healing, it is not surprising that several of the growth factors released from activated platelets, including epidermal growth factor, platelet-derived growth factor, and transforming growth factor β (TGFβ), stimulate proliferative, migratory, and invasive changes in tumor cells [27]. It has been suggested that platelets may play a role in the development of cancer metastases by promoting EMT via TGFβ/Smad and nuclear factor ĸB [28]. Platelet aggregation around tumor cells is thought to aid in tumor survival by protecting the tumor cells from shear forces inside blood vessels and avoiding immune system cells—NK cells in particular [26]. In an experimental mouse model, antibody-mediated depletion of platelets prevented metastasis formation [29]. Furthermore, platelets release heparanase and protease, which degrade the basement membrane, enabling trans-endothelial tumor cell migration into the brain parenchyma [27]. In a study of integrative analysis of the cerebrospinal fluid metabolomic and proteomic profiles of leptomeningeal metastases, the platelet activation and coagulation cascade was a common pathway marked by highly expressed molecules in leptomeningeal metastasis [30].
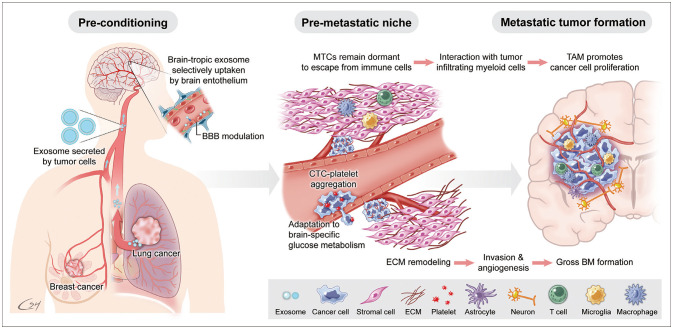
PRECONDITIONING OF METASTATIC NICHE AND METASTATIC LATENCY
The pre-metastatic niche is a necessary home for blood-borne DTCs before proliferation into metastatic tumors (Fig. 1). Kaplan et al. [31] demonstrated that bone marrow-derived hematopoietic progenitor cells that express vascular endothelial growth factor receptor 1 (VEGFR1; also known as Flt1) are home to tumor-specific pre-metastatic sites and form cellular clusters before the arrival of tumor cells. The removal of VEGFR1 cells from the bone marrow of wild-type mice using antibodies abrogates the formation of these pre-metastatic clusters. Hoshino et al. [32] verified that tumor-derived exosomes showed organ-specificity and fused preferentially with the resident cells of various organs, such as the lungs, liver, and brain. In their study, secreted exosomes underwent selective uptake by CD31+ endothelial cells and modulated the brain vasculature to prepare the pre-metastatic niche. Exosome proteomics revealed distinct integrin expression patterns in which the exosomal integrin beta 3 (ITGβ3) was present in brain-tropic cells.
Clinically, tumor recurrence after years of disease-free status after primary treatment, including adjuvant therapy can cause desperation in both the patient and their clinicians. Dormancy of once DTCs in the metastatic niche has been suggested as a precursor to this recurrence, and studies have revealed evidence of stem cell quiescence, extracellular and stromal microenvironments, autophagy, and epigenetics as mechanisms of the dormancy [33]. Malladi et al. [34] suggested that a minority of disseminated cancer cells survive in a latent form for up to years. The latency and immune evasion of metastatic cancer cells are controlled by autocrine WNT inhibition, which imposes a slow-cycling stem cell-like state and enables down-regulation of cell-surface innate immune sensors. The authors verified that latent cancer cells are enriched with SOX transcription factors, which in turn inhibit WNT through DKK1, and persist long-term by evading NK cell-mediated immune surveillance.
METABOLIC ADAPTATION OF BM IN THE BRAIN MICROENVIRONMENT
In a normal brain, glucose is an essential neuronal fuel as the BBB limits penetration of naïve molecules and selectively uptakes essential nutrients via an active transport system. In gliomas, brain tumor-initiating cells compete for glucose uptake by co-opting the high-affinity neuronal glucose transporter, type 3 [35]. In contrast to the Warburg effect, BM cancer cells should enhance aerobic glycolysis via mitochondria to adapt glucose oxidation–dependent brain metabolism. However, persistent metabolism of glucose to lactate is observed in cancer cells, even in aerobic conditions, as an adaptation to intermittent hypoxia. Up-regulation of glycolysis leads to microenvironmental acidosis, requiring evolution to phenotypes resistant to acid-induced cell toxicity [36]. Cell populations that emerge from this evolutionary sequence have a powerful growth advantage as environmental acidosis also facilitates invasion through degradation of the extracellular matrix and promotion of angiogenesis. The proteins upregulated in the BM cells show three major changes in energy metabolism: enhanced glycolysis, increased beta-oxidation of fatty acids, and an elevated pentose phosphate pathway [37]. The cells’ metabolic changes reflect their adaptation to the brain TME, where constant high energy demand is met mostly by glucose oxidation [37]. The authors also found that activation of AMP-activated protein kinase—a key regulator of cellular energy homeostasis that turns on ATP-generating metabolic pathways (e.g., fatty acid oxidation and glycolysis) to preserve ATP levels for cell survival—was observed significantly compared with parent cancer cells and bone metastasis.
INTERACTION BETWEEN CANCER CELLS AND THE BRAIN MICROENVIRONMENT
DTCs should interact with their microenvironment in the brain to establish BMs. The composition of the TME varies depending on the tumor site. The brain TME consists of numerous specialized cell types, such as microglia, astrocytes, and brain endothelial cells [38]. EMT is a well-known mechanism used by cancer cells to achieve distant metastasis. However, EMT explains only half the journey from primary cancer to BM as the tumor cells degrade the extracellular matrix and reach systemic circulation. DTC evolution within the TME might be more difficult in the brain than in other secondary organs because of the inhospitable nature of the naïve organ for incoming cancer cells [39]. It is unknown whether the proteolytic activity of metastatic tumor cells also works in penetrating the BBB. However, several studies have suggested that the high expression of proteolytic enzymes in primary cancer, such as hyaluronidase, cathepsin, and serpins, correlated with increased incidence of BM [14404142]. Neurotrophin is one candidate for helping cancer cells penetrate the BBB because it regulates heparinase, which, in turn, degrades heparin sulfate glycoprotein enriched in brain tissue. In vitro studies revealed that cancer cells attach to brain microvascular endothelial cells and modify the BBB to make it more easily penetrable [43]. Dissociation of pericytes and astrocytes from the vessel wall can also be observed in BMs. As the metastatic cancer cells grow, appropriate angiogenesis is an essential step for mass formation. The brain is one of the most densely vascularized organs, and BMs are among the best-vascularized tumors in humans [38]. Once BM foci are established, cancer cells secrete VEGF to promote angiogenesis, induce peritumoral edema generated by the increased permeability of tumor-associated endothelial cells, and facilitate the leakage of proteins and water into the brain parenchyma surrounding the tumor. Schwartz et al. [44] established a transplantable model of spontaneous melanoma BM in immunocompetent mice. Through transcriptome analysis and intracranial coinjection of melanoma cells with astrocytes, they observed that astrocytes played a functional role in facilitating the initial growth of melanoma cells and suggested that astrogliosis physiologically instigated as a brain tissue damage response is hijacked by tumor cells to support metastatic growth. Zhang et al. [12] substantiated a mechanism of tumor-promoting action by reactive astrocytes for BM development. Using patients’ tumor samples, the authors showed that cancer cells lose PTEN expression after dissemination to the brain compared with those in matched primary tumors but not following dissemination to other organs. They established an experimental mouse model and found that PTEN suppression only occurred when tumor cells were co-cultured with astrocytes. Moreover, inhibition of selected microRNA and exosome secretion by astrocytes resulted in decreased BM growth. This suggests that astrocyte-derived exosomes mediate an intercellular transfer of PTEN-targeting microRNAs to metastatic tumor cells for outgrowth. Furthermore, adaptive PTEN loss in brain metastatic tumor cells leads to increased secretion of the chemokine CCL2 (also referred to as monocyte chemoattractant protein-1), which recruits IBA1 (ionized calcium-binding adaptor molecule 1)-expressing myeloid cells that reciprocally enhance the outgrowth of brain metastatic tumor cells via enhanced proliferation and reduced apoptosis.
IMMUNE MODULATION BEFORE TUMOR PROLIFERATION
In general, the brain is an immune-privileged sanctuary for DTCs escaping systemic chemotherapy. For example, in NSCLC, lower PD-L1 expression and less CD8+ T-cell infiltration were found in BM compared with matched primary tumors, suggesting an immunosuppressive microenvironment in the brain [45]. Reactive astrocytes and tumor-associated macrophages (TAM) are paramount in NSCLC BM and may promote the tumor progression and immune evasion. Once the tumor is formed, various myeloid cells infiltrated. The described cell populations in BM include microglia, TAM, myeloid-derived suppressor cells, Tie2-expressing monocytes, and CD11b+CD45+ vascular modulatory cells [38]. Among these, macrophages are multifunctional cells, and their phenotype is modified by the interaction between the tumor and local environment. The M1/M2 polarization paradigm divides macrophages into 1) those that are activated by the Th1-type cytokines interferon-γ and lipopolysaccharide, resulting in up-regulation of nitric oxide synthase 2 and the pro-inflammatory phenotype (classical activation, M1 macrophages); and 2) those activated by the Th2-type cytokines IL-4 and IL-13, resulting in up-regulation of arginase 1 and leading to pro-angiogenic and pro-tumoral activity (alternative activation, M2 macrophages). Macrophages in the majority of tumors investigated so far seem to have an M2-like phenotype. T cells are another major component of tumor-infiltrating immune cells. Studies have found differences between the T-cell subsets associated with primary cancers and BMs. Mansfield et al. [46] observed an interesting phenomenon: the so-called contraction of T-cell clones in BM compared with paired primary cancer in NSCLC patients. The authors observed decreased numbers of T-cell clones and an expansion of the 10 most abundant T-cell clones in BM compared with primary lung cancers despite the higher mutation burden observed in BM. As such, these results may have implications for immunotherapy. In melanoma BM, Qiao et al. [47] visualized the activation and recruitment of microglia/macrophages during BM development in a mouse model through long-term intravital imaging equipped with bilateral cranial windows. Through transcriptional analysis, they found that microglia/macrophages highly expressed matrix metalloproteinase 3 (MMP3), which was strongly correlated with microglia/macrophage activation and a decrease in ZO-1. In their mouse model, an MMP inhibitor moderately decreased the occurrence of melanoma BM, which suggests that MMP3 secreted by microglia/macrophages may facilitate melanoma cell growth.
A recent study by Gonzalez et al. [48] identified a distinctive subset of tumor-infiltrating lymphocytes in BMs of various primary cancers using single-cell transcriptomics and high-dimensional mass cytometry (cytometry by time-of-flight, Cy-TOF). The authors performed an integrative analysis of over 100,000 malignant and non-malignant cells from 15 human BM tissues and revealed stromal immunosuppressive states enriched with infiltrated T-cells and macrophages. They found two macrophage states: 1) MAMs:APOE+, associated with therapy resistance and angiogenesis; 2) MAMs:S100A8+, associated with immunosuppression. They also suggested that there were common denominators among BM cancer cells from various primary cancers, which they classified into two architectures—highly proliferative and highly inflammatory—in a framework using eight functional metaprograms of the transcriptome. The authors’ analysis of the tumor-stroma interface found that T-cell anergy correlates with BM cell proliferation.
CONCLUSION
BM penetrates the BBB not simply by chance but by selective organotropism of primary cancer cells, supported by both clinical observations of the varying proportional incidence of BM among primary cancers and experimental evidence of cancer cell tropism for specific “soil” organs. Clonal selection of primary cancer cells and acquired somatic mutation during BM processes offer clues for this brain organotropism. The concepts of the pre-metastatic niche and the dormant stage before tumor cell proliferation support the fact that DTCs struggle to survive and proliferate in the brain’s relatively fertile TME and require time and complex processes to modulate the TME, including infiltrating immune cells appropriate for tumor mass formation (Fig. 2). This wise adaptation of DTCs to the brain TME seems to make them invincible to be inhibited by one or a few targeted therapies. However, the recent advancement of single-cell analysis reveals possible common denominators among BMs from various primary cancers, and detailed immune cell subset profiles seem to help identify ways to stop the immune evasion of key regulators of BM cancer cells.
 XML Download
XML Download