

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Meningioma is the most common primary brain tumor in adults, comprising 35.8% of all primary central nervous system (CNS) tumors and more than 53% of all benign CNS tumors, with an even higher prevalence in autopsy and imaging studies. From health screening data, meningiomas affect almost 3% of women. Meningioma arises from progenitor cells: 1) arachnoid cap cells of the arachnoid layer and 2) fibroblasts in the inner dura mater. Although most meningiomas can be treated with complete excision, it is often difficult to achieve gross total surgical resection and is life-threatening. In the histopathological diagnosis criterion for atypical meningioma, which is different from grade 1 typical meningioma, brain invasion was clarified as the standard-alone criterion for atypical meningioma in the World Health Organization (WHO) 2016 CNS tumor classification [1]. Whether brain invasion is a significant factor in relapse remains controversial [23456]. However, in a large series of meningiomas of all grades, brain invasion in recurrent atypical meningiomas has been reported to increase the risk of recurrence compared with non-atypical meningioma [7]. Brain-invasive meningioma has a high recurrence rate, making it difficult to resect completely, increasing patient mortality and making treatment challenging [8]. If there is a residual tumor after surgery, the risk of recurrence is high [9]. Whether adjuvant radiation therapy reduces the risk of recurrence after total resection in grade 2 meningiomas is unclear [10]. However, the failure of local accommodation or radiation-related neurological deficits after radiation therapy is a frustrating factor for clinicians. Although some authors have attempted pharmacotherapy for brain-invasive meningioma patients with surgical inaccessibility or radiotherapy, the role of pharmacotherapy in meningioma is unclear, and no clinical trials have yielded significantly positive results [10].
As next-generation sequencing is being applied in clinical practice, the understanding of the molecular biology of invasive brain meningiomas is expanding. In addition, advances in cancer genomics have greatly improved our understanding of molecular alterations in the 2021 WHO classification. Some of these findings have important implications for tumor classification, patient care, and the design and interpretation of clinical trials [11].
Herein, we aimed to understand the molecular mechanisms of brain invasion in atypical meningiomas and review the genetic background involved. Thus, the molecular understanding of invasive meningioma has improved, various potential treatments have been introduced, and research trends have been reviewed.
BRAIN INVASIVE MENINGIOMA
In the 2016 WHO classification of CNS tumors, brain invasion became the sole criterion for grade 2 meningioma, as was the criterion for mitosis of ≥4 per 10 high-power fields [12]. In addition, a diagnosis can be made if three of the following five additional diagnostic criteria are met: spontaneous necrosis, sheeting (loss of whorling or fascicular architecture), prominent nucleoli, high cellularity, and small cells (tumor clusters with a high nuclear: cytoplasmic ratio) [12]. With the addition of this brain invasion-only criterion, brain-invasive otherwise benign, which was previously classified as grade 1, has been classified as grade 2 in atypical meningioma since the WHO classification in 2016. Brain invasion meningioma causes loss of interface with brain tissue on T2 weighted magnetic resonance imaging and causes prominent peripheral brain edema. Even in microscopic surgery, it penetrates the arachnoid interface or encases cerebral blood vessels [8]. An important structure that supports or prevents brain involvement is the so-called “brain-meningioma interface.” It can be divided into three types: bone invasion, perivascular growth along the Virchow-robin-spaces, and direct brain invasion [13].
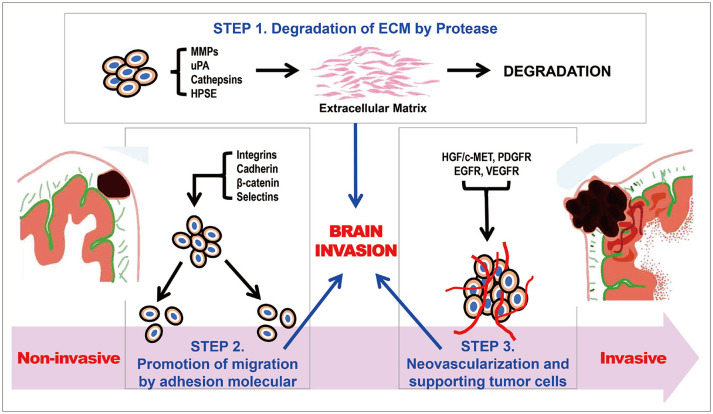
Major process of meningioma brain invasions in three steps
Degeneration of extracellular matrix (Fig. 1)
First step is the degeneration of the extracellular matrix (ECM) by protease [8]. The ECM and basement membrane (BM) are rigid structures formed from macromolecules: type IV collagen, laminin, entactin, nidogen, fibronectin, and heparin sulfate proteoglycans (HSPG). Proteases can degrade BMs and connective tissues. Proteases result in a breakthrough interaction between the ECM and BM. Matrix metalloproteinases (MMPs), one of the proteases, are lysosomal endopeptidases that are categorized into four classes: category I, interstitial collagenases (MMP-1, MMP-8, and MMP-13), which degrade fibrillary collagens; category II, type IV collagenases (MMP-2 and MMP-9), which degrade the BM collagens gelatin and elastin; category III, stromelysins (MMP-3, MMP-10, and MMP-11), which degrade proteoglycans, fibronectin, laminin, gelatin, and globular proteins of type IV collagens; and category IV, membrane-type MMPs (MMP-14, MMP-15, MMP-16, and MMP-17), which contain a unique transmembrane domain in their C-terminus, localizing these MMPs to the cell surface. Jalali et al. [14] confirmed that MMP-16 knockdown tumors showed reduced invasion into brain tissue. In their experiments, they confirmed that MMP-16 expression was significantly elevated in bone- and brain-invasive meningiomas and that the number of cells passing through the BM and the mobility of knockdown cells were decreased [14]. MMP-16 is believed to promotes invasiveness of meningiomas [14]. Meningiomas may have a wide range of epithelial and mesenchymal properties, including the ability to produce collagen and other ECM proteins [15]. Immunoreactivity to MMP-2 and MMP-9 and their tissue inhibitors TIMP-1 and adhesion factor galectin-3 were found in most cases of benign meningiomas regardless of location [15]. This suggests that these compounds may serve as potential therapeutic targets.
Another potential target of MMP inhibitors, uPA and PAI-1, is tumor invasion. Kandenwein et al. [16] confirmed a significant correlation between WHO grade and the concentrations of uPA and PAI-1 proteins in meningiomas. In that study, uPA/PAI-1 levels were significantly increased in high-grade meningiomas and showed an inverse correlation between their expression levels and promoter methylation. In addition, brain involvement was frequent in tumors with higher PAI-1 expression (more frequent in tumors containing >6 ng/mL PAI-1), although the Simpson grade was not significant and there was a highly significant correlation with patient prognosis [16].
Lah et al. [17] reported that cysteine cathepsins and their inhibitors were involved in the early recurrence of meningiomas, regardless of histological classification. Lysosomal cysteine proteinases such as cathepsin D and cysteine cathepsins B and L initiate a proteolytic cascade by activating cysteine protease [17]. Subsequently, lysosomal cysteine proteinases activate cell membrane-associated pro-urokinase, inducing the extracellular release of plasmin from plasminogen, and finally activate various types of MMPs which degrade collagen and other basal lamina proteins [17].
HSPG act as cellular switches to invasive meningiomas [18]. HSPG, a core protein of the ECM BM, and heparanase (HPSE), a major enzyme that degrades heparin sulfate, are important regulators of the ECM [18]. HPSE breaks the heparin sulfate chains of HSPG into 5–7 kD-sized fragments at specific HSPG sites [18].
Promotion of tumor cell migration to resident cells by adhesion molecules (Fig. 1)
Integrins are cell-surface adhesion molecules that are important for proliferation and migration. Recently, much interest has focused on the role of integrins in carcinomas. Each histological meningioma subtype has a specific spectrum of integrin expression, especially αVβ3 and αVβ5 integrins, which contribute to the invasive growth of meningiomas. αvβ5 is an integrin heterodimer that is predominantly expressed in meningiomas, whereas αvβ3 is mainly detected in tumor vessels [19]. Brain invasion is inhibited in meningioma cell lines and mouse models injected with cilengitide [19]. Integrins along with their inhibitor, cilengitide, could be a potential targeted therapy.
E-cadherin, a cell-surface glycoprotein, is essential for calcium-dependent cell–cell adhesion and structural rigidity. The extracellular N-terminus of E-cadherin forms a “zipperlike” structure that serves as a tight junction [20]. Additionally, the intracellular part is involved in the construction of the cytoskeleton indirectly to the cell–cell junction by catenin (α-catenin and β-catenin). Disruption of these junctions causes brain invasion, and the expression of E-cadherin and β-catenin is closely related to the meningioma grading criteria [20].
Selectin is a key protein involved in this process. Selectin mainly correlates with the binding, rotation, and extravasation of activated leukocytes, which commonly occurs on the endothelium and in inflammatory reactions. E-, P-, and L-selectins in turn have the following functions: E-selectin secretion is activated following local stimulation by endothelia of skin and bone marrow and subsequently induced by inflammatory cytokines, P-selectin exocytosis translocates to the cell surface of activated endothelial cells and platelets, and L-selectin functions by interacting with the selectin binding ligand (P-selectin glycoprotein ligand-1). Atukeren et al. [21] confirmed that these selectins are expressed at higher levels in meningiomas than in the control group, suggesting that selectins may be involved in the pathological mechanism of meningiomas.
Neovascularization and supporting meningioma tumor cells by growth factors (Fig. 1)
Growth factors play critical roles in brain invasion processes, including the promotion of migratory, proliferative, and angiogenic responses in meningioma cells [8]. The hepatocyte growth factor (HGF)/c-MET signaling pathway is one of the major pathways involved in this process. HGF is a multifunctional growth factor secreted by mesenchymal cells and has a strong mitogenic effect on hepatocytes. It consists of a heavy chain with four domains and a light chain, and binds to its tyrosine kinase receptor (RTK), a product of the proto-oncogene c-MET [22]. Mature c-MET is structurally distinct from most RTKs. It exists as a heterodimer containing an extracellularα-chain and transmembrane β-chain. Once activated by HGF binding, c-MET is auto-phosphorylated and recruits adaptor proteins, activating multiple downstream effector proteins and signaling cascades [22]. Dysregulation of the HGF/c-MET signaling pathway induces tumor cell proliferation, motility, and invasiveness in several human cancers, including breast cancer, lung cancer, and hepatocellular carcinoma (except for clinical trials in meningioma) [22].
Platelet-derived growth factor (PDGF) and endothelial growth factor receptors (EGFRs) (Ras/Raf/MAPK and PI3K pathways) are also potential therapeutic targets. PDGF acts as a proliferation driver during normal development and in multiple cancers, and all histological grades of meningiomas express PDGF ligands AA and BB [23]. Early studies have shown that normal arachnoid cells almost exclusively express β-type PDGF receptors [2425], which predominantly bind to PDGF-BB and stimulate meningioma growth, activate MAPK, and induce c-Fos expression [2627].
More than 60% of meningiomas highly express the EGFR [28]. EGFR activation by autocrine or paracrine mechanisms in meningiomas may promote tumor growth [8]. EGFR activation increases resistance to apoptosis, promotes angiogenesis, and impairs immune surveillance. Thus, EGFR inhibitors have the potential to reduce tumor progression [8].
EGF and transforming growth factor-α (TGF-α) activate their receptors, which promote the proliferation of meningioma cells in an in vitro study [29]. In addition, upregulation of TGF-α activity in meningioma and tumor specimens has been demonstrated to be associated with aggressive growth [29]. Signal transduction activated by RTK, including PDGF receptor (PDGFR) and EGFR, is mediated by Ras/Raf/MAPK and PI3K pathways [30]. Imatinib is an oral tyrosine kinase inhibitor that targets BCR-ABL, PDGFR, and c-Kit receptors [8]. Meningiomas frequently overexpress PDGFR-α and PDGFR-β, which could be potential targets for imatinib therapy [8].
Vascular endothelial growth factor (VEGF) and the VEGF receptor (VEGFR) are highly expressed in meningiomas [31]. This is related to the extent of peritumoral edema. Moreover, the strong correlation between meningioma grade and VEGF suggests that anti-angiogenic therapy may be beneficial for the treatment of invasive meningiomas. An anti-VEGF monoclonal antibody (bevacizumab) improves the survival of several malignancies, including those of colorectum, lung, breast, and glioblastomas [3233]. This has been studied in several retrospective analyses and in two phase II trials involving patients with refractory meningiomas [34]. Sunitinib, a wide-spectrum tyrosine kinase inhibitor, targets VEGFR, PDGFR, and several other oncogenic pathways and is currently being used in clinical practice for several cancers [31].
Promising treatment for brain-invasive meningiomas
Immunotherapy
A recent study revealed reduced expression of PD-1-expressing T lymphocytes, such as CD4+ and CD8+, in anaplastic meningiomas [35]. Immunotherapy using PD-1/PD-L1 has been approved by the United States Food and Drug Administration; however, much remains unknown about the meningioma microenvironment [8]. However, recent clinical trials are ongoing, and current immunotherapy strategies for meningioma, anti-PD1 drugs pembrolizumab, and nivolumab are undergoing clinical trials for recurrent or residual high-grade meningiomas [8].
Depending on the grade of meningioma, the expression pattern of PD-L1 is different [35]. In grade 1 meningiomas, the overall expression level of PD-L1 is very low, and CD68 is expressed in a small number of cells. In grade 2 meningiomas, a significant PD-L1+population shows CD68+ expression, in addition to a separate population with CD68+/PD-L1–cells. In grade 3 meningiomas, PD-L1 expression is predominant in CD68 cells.
Hormone therapy
The epidemiological incidence of meningioma is high in women, particularly during pregnancy and breast cancer [3637]. The functions of progesterone and estrogen can be considered as regulatory mechanisms and potential therapeutic targets.
In a large-scale study, estrogen-alone hormonal replacement treatment (HRT) in postmenopausal women increased the incidence of meningioma, but not with estrogen plus progesterone HRT [383940]. Additionally, HRT without oral contraceptives plays an important role in meningioma formation [41].
Progesterone receptors are found in 58%–83% of meningiomas, but estrogen receptors are found in only 0%–8% [42]. Therefore, since meningiomas have a higher expression rate of progesterone receptors, they may be potential therapeutic targets for growth factor inhibition [8]. However, a recent double-blind phase 3 clinical trial using mifepristone, an anti-progesterone drug, failed to control unresectable meningioma [43].
Tumor treating field
Tumor-treating fields (TTFs) are antimitotic treatments that selectively affect dividing glioblastoma cells by delivering a low-intensity, medium-frequency (200 kHz) alternating electric field through transducer arrays applied to the scalp, and their safety and effectiveness have been reported [444546]. In the same way, it can be considered that TTF is applied to meningioma cells to interfere with the mitosis process and induce violent membrane blebbing, which subsequently induces immunogenic cell death [8].
Wafers
Wafer treatment is a method of integrating chemotherapeutic agents into biodegradable polymers and continuously releasing them locally. The results of a placebo-controlled trial of wafer treatment using biodegradable polymers impregnated with carmustine for recurrent malignant gliomas have been reported [47]. Therefore, potentially biodegradable copolymers can be considered as impregnated chemotherapies for brain-invasive meningiomas.
GENETIC LANDSCAPE OF MENINGIOMA
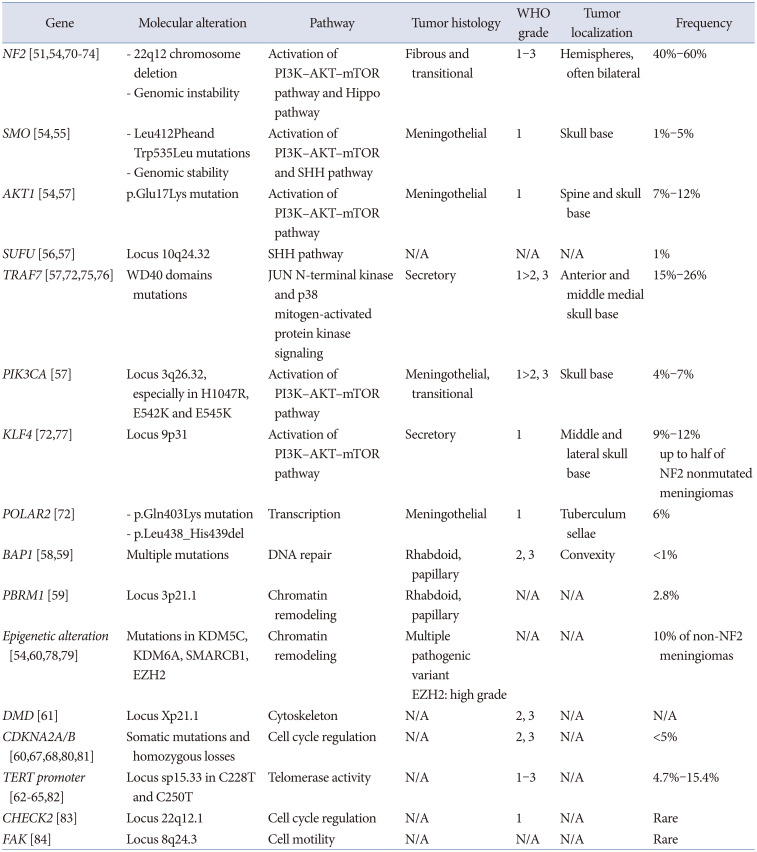
The histopathological classification of meningiomas is divided into three grades based primarily on histologic features. Each grade has different morphological characteristics and is divided into various histological subtypes. A low-grade meningioma may recur despite appropriate surgical excision or a high-grade meningioma may not recur. This fact indicates that the existing grading system based on histological characteristics does not consistently predict the natural history of each meningioma [48]. Further understanding and insight into genomics offers the potential for tumor grade-based natural history prediction and targeted therapy for meningioma [48]. Common molecular alteration is summarized in Table 1.
Somatic mutational profile of sporadic meningiomas
Among sporadic meningioma cases, 80% are based on specific somatic driver mutations [49]. They include the following variants: 1) neurofibromatosis-2 (NF2) with or without co-mutation in SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1 (SMARCB1); 2) mutations in the WD40 region of TNF Receptor-associated factor 7 (TRAF7), which can occur alone; 3) with a recurrent mutation in Kruppel-like factor 4 (KLF4K409Q); 4) mutations in PI3K (phosphoinositide 3-kinase) pathway molecules, including phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), PIK3R1, and AKT1E17K; 5) Hedgehog (HH) signaling molecules (i.e. SMO, suppressor of fused [SUFU], PRKAR1A); 6) recurrent mutations in RNA Polymerase II subunit A (POLR2AQ403K or L438_H439del); or 7) SMARCE1 mutations [49].
The most common driver mutation is NF2 biallelic loss, which accounts for approximately half of meningiomas [4850]. Mutations in grade 1 meningiomas include TRAF7 (28%), AKT1 (14%), KLF4 (12%), and SMO (5%), which occur in a mutually exclusive pattern with NF2, accounting for approximately 20% [48].
Genomic analysis alteration in intracranial meningiomas
Mutations caused by deletion of the 22q12 chromosome encoding the tumor suppressor gene Merlin (also called neurofibromin 2, NF2) occur in approximately half of meningioma cases and are associated with the fibroblastic/transitional subtype [9]. Merlin coordinates membrane receptor signaling and cell-to-cell contact, and is a critical regulator of contact-dependent inhibition of proliferation [51].
NF-2 gene alteration
NF-2 inactivation results in genomic instability and peculiar multiple localizations in the hemispheres [9]. NF-2 mutant meningiomas have larger volumes, fibrous or atypical histology, male predominance, and preferentially occur along the cerebral convexities posterior to the coronal suture. Along the skull base, they show lateral to medial gradient, originating along the lateral sphenoid wing, invading the bone [52]. SMARCB1/NF2 co-mutations localize anterior to the coronal suture and medially along the falx [53].
Non-NF2 gene alteration
SMO and v-akt murine thymoma viral oncogene homolog 1 (AKT1) can lead to activation of the PI3K–AKT–mTOR pathway, which is associated with meningiomas localized in the skull base and with genomic stability [5455]. SUFU and patched 1 mutations can activate the SHH pathway, and are found in 1% of sporadic meningiomas [56].
TRAF7 mutations are found in approximately 25% of grade 1 and 2 meningiomas and can be associated with alterations in AKT1, KFL4 or PIK3CA alteration [57].
Mutations in two hotspots of RNA polymerase II subunit A (POLR2A) are associated with the meningothelial subtype, and meningiomas with these mutations are usually located in tuberculum sellae [9].
Inactivation of breast cancer-associated protein 1 (BAP1) is frequent in rhabdoid meningiomas and appears to be associated with early tumor recurrence [58]. Biallelic inactivation of polybromo 1 (PBRM1) has been reported in papillary meningiomas [59].
Upregulation of enhancer of zeste homolog 2 is found in atypical meningiomas and appears to be a marker of aggression and higher grades [60]. Somatic deletion of the Duchenne muscular dystrophy gene was also found in meningiomas and appears to be associated with a poor prognosis [61].
Mutations in KDM5C (lysine demethylase 5C), KDM6A or Somatic SWI/SNF-related matrix associated actin-dependent regulation of chromatin subfamily B member 1 protein (SMARCB1) [54].
Genetic markers for the potency of malignancy
A telomerase reverse transcriptase (TERT)-promoting mutation is present in secondary atypical meningiomas that progress from low-grade primary tumor [6263]. Several reports have shown that the TERT promoter mutation is associated with poor prognosis and shorter overall survival (OS), regardless of the WHO grade of meningioma [6465].
The MLL/KMT2 mutation found in WHO grade 2 chordoid meningioma is associated with NF2 mutations and poor prognosis [66].
Cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) is a tumor suppressor. In meningiomas, this mutation can progress the tumor from grade 2 to grade 3 [6067]. The overexpression of CDK6 and pRB proteins has been shown to be predictive of relapse [68]. CDKN2A alteration (p.Ala148Thr) mutation, whole homozygous or heterozygous gene loss, or promoter methylation strongly correlated with recurrence and with a Ki67 labeling index >7% [69].
Copy number alterations in intracranial meningiomas
Copy number alterations (CAN) are associated with the risk of recurrence after resection of atypical meningiomas and can be used as an index for adjuvant radiation therapy decisions [57]. CANs include the most common 22q deletion and 1p, 14q, and 9p losses associated with higher-grade meningiomas [85], with 6q, 10q, and 18q loss or the gain of 1q, 9q, 12q, 15q, 17q, 19q, 20q, and 5 chromosomes [4878]. Previous studies have reported that co-deletion of DNA repair and tumor suppressor gene, CHEK2, is frequently found with NF2 on chromosome 22q and promotes chromosomal instability [83].
EPIGENETIC ALTERATION IN MENINGIOMAS
Epigenetics can be defined as mitotically heritable changes in gene expression that are not caused by changes in the primary DNA sequence. Epigenetic mechanisms, including those involving enzymatic modifications of DNA or histone proteins, thereby regulating gene expression, are increasingly recognized as a source of phenotypic variability in biology. In addition to DNA methylation and histone modifications, other potential epigenetic mechanisms include specific deposition of histone variants, non-coding RNA chromatin remodeling, nuclear organization of RNAs, chromatin remodeling, and nuclear organization of DNA. The interplay between histone modifications and other chromatin modifications leads to dynamic regulation of chromatin structure, thereby affecting several relevant cellular processes, including transcription, DNA replication, DNA repair, and genomic stability. This epigenetic regulation can also play a major role in meningioma tumorigenesis (Table 2).
Methylation profiles in intracranial meningiomas
Sahm et al. [86] classified six methylation clusters in relation to typical mutational, cytogenetic, and gene expression patterns in 497 meningiomas collected at ten European academic neuro-oncology centers, using an alternative methylation chip. Classification according to these DNA methylation classes accurately identifies patients with high progression risk in WHO grade 1 tumors and low risk of recurrence in grade 2 tumors than that based on histology (p=0.0096 from the Brier prediction test) [86]. They are classified into group A, which contains four sub-methylation clusters (ben-1, ben-2, ben-3, and int-A), and two sub-methylation clusters (int-B, mal), for a total of six methylation clusters [86].
Olar et al. [87] also presented confirmation results to interpret the clinical significance of meningiomas and methylation profiles. They included a final optimized 64-CpG loci meningioma methylation predictor (64-MMP) stratified clustering, using 283 probes from 51 validated datasets. They were classified into two methylation groups that were significantly different (p=0.0003, log rank) in survival [87]. In this study, there were two subgroups (prognostically favorable vs. unfavorable molecular methylation subgroups). The prognostically unfavorable subgroup presented a higher proportion of copy number aberrations, including losses of 1p, 6q, 14q, and 18q, and a gain of 1q, all associated with poor outcomes, with 185 hypermethylated CpG loci and a median recurrence-free survival (RFS) of 12.07 years (range, 0.31–17.61 years) [87]. The favorable subgroup presented 98 hypermethylated CpG loci, which did not reach median RFS (range, 0.27–16.6 years) [87].
Similarly, Nassiri et al. [88] also reported that DNA copy number analysis demonstrated an increased frequency of copy number aberrations in the higher risk groups due to a high proportion of chromosomal deletions in 1p, 4p, 6q, 10q, 14q, and 18q. They were divided into groups according to WHO grade, and there was a significant difference in median RFS in the high-risk group versus the low-risk group within the groups (Grade I: hazard ratio [HR] 2.9, 95% confidence interval [CI]: 1.3–6.6, p=0.006; Grade II: HR 2.8, 95% CI: 1.7–4.8, p<0.001; Grade III: HR 3.0, 95% CI: 1.4–6.5) [88]. They created a freely available online calculator of the meningioma recurrence score to facilitate unrestricted global dissemination [88].
Correlation of meningioma relevant mutations with methylation classes
Correlations were identified between mutations in meningioma and methylation classes, and the TRAKL mutation genotype was significantly more frequently observed in benign MC (62.5%) than in other MC types (p<0.001). KLF4 and TRAF7 mutations were also more common among benign MC (39.1% and 59.4%, p<0.001) [89]. Consequently, the TRAKL mutation genotype was more common in benign MC (62.5%, p<0.001) [89]. NF2 mutations were significantly more frequently observed in malignant MC (50.0, p<0.001) [89]. Furthermore, TERT promotor mutations were more frequently observed in malignant MC (11.1%) than in benign (0%) and intermediate MC (4.5%; p<0.04) [89].
Basics of epigenetics and histone modification
Chromatin and nucleosome
Chromatin is a condensed combination of DNA and histones within the cell nucleus. The structural and functional unit of chromatin is a nucleosome, which consists of a discshaped octamer composed of two copies of each histone protein (H2A, H2B, H3, and H4), around which 147 base pairs of DNA are wrapped twice [90]. Electron microscopy studies have revealed that the organization of nucleosomal arrays structurally resembles a series of “beads on a string,” with the “beads” being the individual nucleosomes and the “string” being the linker DNA [91]. Methylation is required to silence transposable elements to maintain genomic stability, and is a critical regulator of genes that contribute to cell pluripotency [90].
Transcription activation
Each of these histone modifications directly or indirectly affects the chromatin structure, thereby leading to alterations in DNA repair, replication, and gene transcription. Heterochromatin is condensed into DNA methylated and deacetylated histones, making it inaccessible to transcription factors. Lysine acetylation neutralizes the positive charge on histone tails, weakens histone-DNA or nucleosome-nucleosome interactions, and induces open (euchromatin-like) conformational changes [9293]. This unstable nucleosome and chromatin structure allows other nuclear factors to easily access DNA [90].
Histone modification sites
The methylation of histone residues occurs at the side chains of arginine and lysine. Histone H3 is primarily methylated at four lysine residues within the N-terminal tail (K4, K9, K27, and K36) [94]. Histone modification is regulated by histone 3 lysine methyltransferase and histone 3 lysine demethylase [90].
Loss of H3K27me3 in WHO grade 3 meningioma
In WHO grade 3 meningioma, loss of H3K27me3 distinguishes meningioma patients with an unfavorable prognosis; however, it has not yet been established as a prognostic biomarker in WHO grade 3 meningioma [95]. Maier et al. [95] reported H3K27me3 status as complete loss, <50%, and >50% stained cells in 110 tumor samples from a population-based consecutive cohort of 40 WHO grade 3 meningioma patients. However, they found no difference in OS in patients with >50% H3K27me3 retention compared to <50% in a cohort of patients with WHO grade 3 meningioma (Wald test p=0.5) [95]. H3K27me3 expression differed without a discernible pattern between biopsies from repeated surgeries for meningioma recurrences [95]. Loss of H3K27me3 may be a general tendency for aberrant methylation in more aggressive tumors [96]. However, it is not compatible with a systematic pattern of immunohistochemical H3K27me3 loss associated with OS or malignant transformation of meningiomas [96]. Moreover, it did not support H3K27me3 loss as a useful immunohistochemical biomarker for grade 3 meningiomas because of staining-specific challenges in quantification [95].
Histone modification regulating apoptosis in atypical meningioma
Previous studies have shown that histone protein modification enzymes are epigenetically associated with recurrence of atypical meningiomas. Overexpression of apoptosis-associated factors and histone-modifying enzymes in the immunohistochemical analysis showed that certain apoptosis-associated factors should be associated with recurrence of atypical meningiomas, which may be regulated epigenetically by histone-modifying enzymes [94]. Multivariate analysis showed immunohistochemically low expression of CASP3 (HR 5.243, p<0.001) and BAX (HR 6.338, p<0.001), and immunohistochemical overexpression of survivin (HR 4.415, p=0.007), BCL2 (HR 3.699, p=0.013), and MDM2 (HR 2.946, p=0.037) [94].
Expression of microRNAs (miRNA-21 and miRNA-107) in meningioma
miRNAs are members of a non-coding endogenous RNA region of approximately 22 nucleotides [97]. It has been shown that miRNAs play important roles in many biological processes, including metastasis, proliferation, apoptosis, stress resistance, tumorigenesis, and cell differentiation [9899]. In particular, as the histopathological grade of meningioma increased, the expression of the anti-apoptotic factor miRNA-21 increased, while the expression of the tumor suppressor miRNA-107 decreased significantly [97].
Downregulated miRNA-145 in atypical and anaplastic meningiomas
The miRNA-145 in meningiomas reduces proliferation and increases apoptotic sensitivity. The expression analysis of miRNA-145 and let7a-d revealed significantly lower expression levels of miR-145 and let7d in meningiomas compared with arachnoidal tissue [100]. Significantly lower expression of miRNA-145 in WHO grade 2 and grade 3 meningiomas than in WHO grade 1 meningiomas and pesticidal tissue was observed [100]. In PCR array studies of miR145 overexpressing cells, collagen V alpha (COL5A1) expression was downregulated by miR-145 overexpression [100]. This indicated that collagen type V is a potential target of miR-145. Thus, COL5A1 expression is significantly upregulated in atypical and anaplastic meningiomas [100].
CONCLUSIONS
Increasing knowledge of the molecular landscape of meningiomas has allowed the identification of prognostic and predictive markers that can guide therapeutic decision-making processes and the timing of follow-up. It is important to understand the three major steps for brain invasion of meningeal cells: 1) degradation of ECM by proteases, 2) promotion of tumor cell migration to resident cells by adhesion molecules, and 3) neovascularization and supporting cells by growth factors. The genomic landscape of meningioma should be analyzed by major categories, such as germline mutations in NF2 and somatic mutations in non-NF2 genes (TRAF7, KLF4, AKT1, SMO, and POLR2A). Epigenetic alterations in meningiomas have been studied, focusing on DNA methylation, histone modification, and RNA interference. However, there are difficulties in establishing the epigenetic role of certain genes owing to the frequent interactions among each mechanism.

 XML Download
XML Download