

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Oxygen is indispensable for cellular functions of living organisms. Cells utilize oxygen to generate energy through the conversion of nutrients to adenosine triphosphate (ATP) via an oxygen-dependent pathway known as cellular respiration [1]. An excessive level of oxygen is referred to as hyperoxia, whereas an insufficient level is referred to as hypoxia; an oxygen level within the normal range is termed normoxia. Following the seminal observation by Louis Pasteur that living organisms consume oxygen [2], discoveries by Otto Warburg and Corneille Heymans further elucidated the mechanism by which the protein complex hemoglobin interacts with oxygen [3] and how the central nervous system (CNS) responds to oxygen [45]. Three Nobel Prize recipients proceeded to demonstrate the mechanism by which living organisms sense oxygen levels in the body. In 2019, William G. Kaelin Jr., Sir Peter J. Ratcliffe, and Gregg L. Semenza won the Nobel Prize for their discovery of hypoxia-inducible factor 1-alpha (HIF1A) and its role in oxygen sensing mechanisms [678910]. We now have a better understanding of how cells sense and respond to variations in oxygen levels.
Although the brain comprises only 2% of total body weight, it utilizes 20% of the total oxygen used within the body. Within the brain, the consumption and distribution of oxygen are region-dependent; oxygen levels within the midbrain comprise only 0.5% of total oxygen content, whereas pial oxygen levels comprise approximately 8% [11]. Because oxygen is important for cellular metabolism, tumor cells consume high levels of oxygen. Therefore, brain tumor regions are typically hypoxic, with tumoral and peritumoral regions containing oxygen concentrations of approximately 1.25% and 2.5%, respectively [12]. Hypoxia is a clinical hallmark of many cancers, including brain tumors, and is typically associated with negative outcomes in patients [13]. Multiple factors can contribute to brain tumor hypoxia. The most unique characteristics of tumor cells are their high proliferation rate and metabolic demands. Due to the fast rate of proliferation, tumor tissues can expand a long distance from blood vessels, limiting oxygen diffusion to the center of tumor tissues. In addition, abnormal angiogenesis induced by the tumor microenvironment (TME) results in constriction of blood vessels, further inducing hypoxia [1415].
Hypoxic conditions can affect not only tumor cells, but also blood vessels, stromal cells, and immune cells. Although hypoxia is beneficial for certain processes, including the germinal center (GC) reaction, hematopoiesis, and intestinal barrier maintenance [16], it hinders antitumor immune responses. For example, hypoxia induces the accumulation and immune-suppressing actions of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), thereby reducing the functionality of antitumor immune cells such as CD8 T cells [17]. Therefore, hypoxia induces a vicious cycle of antitumor immune responses and it is important to better understand how hypoxia regulates the TME.
MOLECULAR MECHANISM OF SENSING HYPOXIA
Hypoxia-inducible factor (HIF) is the most well-characterized sensor for oxygen tension [18]. HIF is a transcription factor that is constituted by two distinct subunits, HIFα and HIFβ. Humans express three isoforms of the HIFα subunit (HIF1α, HIF2α, and HIF3α). HIF1α is expressed ubiquitously and is overexpressed in tumor cells, whereas HIF2α expression is restricted to certain cell types such as subsets of tumor-associated macrophages (TAMs). HIF3α expression is also cell-specific, and immune cell expression has not been confirmed [1920]. HIF1α and HIF2α act as transcription factors that target both unique and overlapping sets of target genes. In addition, HIF1α subunits can dimerize with HIF1β, also known as the aryl hydrocarbon receptor nuclear translocator (ARNT), that is ubiquitously expressed. Although HIF3α is a negative regulator of HIF1, it can also function as a transcriptional activator of distinct genes [21].
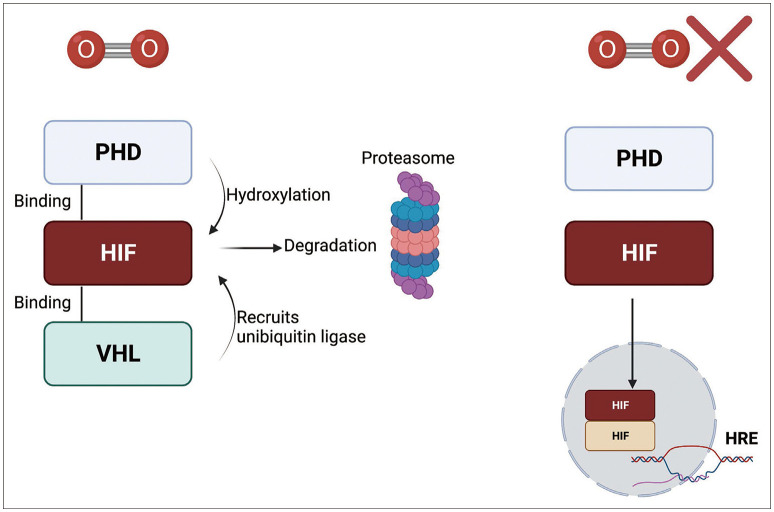
Under normoxic conditions, HIFα is bound to prolyl hydroxylase domain (PHD) proteins 1–3 through an oxygen-dependent degradation domain (ODDD). PHD is sensitive to oxygen levels due to its 2-oxoglutarate and iron-dependent dioxygenase domains, and is therefore affected by changes in oxygen tension [22]. PHD hydroxylates the prolyl residues of HIFα, which induce ubiquitination through E3 ubiquitin ligase interactions with the von Hippel-Lindau tumor suppressor protein (VHL), thereby promoting the proteasomal degradation of HIFα [23]. However, decreases in oxygen tension result in decreased PHD activity and therefore lead to stabilization of HIFα. Stable HIFα proteins translocate to the nucleus, bind to HIF1β and other coactivators, and influence downstream transcription (Fig. 1) [24].
Downstream genes targeted by HIF-mediated transcription are known as hypoxia-response elements (HREs) that are involved in metabolism and proliferation among other cellular functions. For example, hypoxia stabilizes epidermal growth factor receptor variant iii (EGFRviii) by enhancing its interaction with integrin β1 within brain tumor cells. This interaction facilitates the transport of other integrins to the cell surface leading to activation of focal adhesion kinase (FAK) and tumor cell invasion [25]. Hypoxia also represses cap-dependent translation, resulting in the downregulation of global translation and an increase in the selective translation of stress-related proteins [26]. For example, hypoxia-induced activation of inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α) is responsible for activation of the stress-related protein X-box binding protein 1 (XBP1) [2728]. In addition, hypoxia mediates metabolic changes that can lead to mitochondrial dysfunction. Severe hypoxia below an oxygen level of 0.3% disrupts electron transport complex activity [29]. Hypoxia also attenuates the tricarboxylic acid (TCA) cycle, induces mitochondrial fission, and activates mitophagy in both HIF-dependent and HIF-independent manners [303132]. A characteristic feature of hypoxic cells is enhanced glycolysis that is mediated through HIF-induced upregulation in the expression of the glucose transporter, pyruvate kinase M2, lactate dehydrogenase, and phosphoinositide-dependent kinase 1 [3334] and in turn, increased lactate production and low pH levels. In addition, hypoxia regulates the expression of several angiogenesis-related genes, most notably vascular endothelial growth factor (VEGF) as well as placenta growth factor (PGF), angiopoietin, C-X-C motif chemokine ligand 12, and platelet-derived growth factor B [35]. VEGF signaling induces endothelial proliferation, and both VEGF and PGF induce extracellular matrix degradation [3637]. Hypoxia can also regulate the radiosensitivity of brain tumor cells, although the mechanisms are unclear [38].
HYPOXIA THE THE IMMUNE SYSTEM
Like other cells, immune cells need an appropriate level of oxygen that increases with greater activity. Although normoxic air contains 21% oxygen, some tissues may have hypoxic oxygen concentrations despite normal physiological conditions [18], known as “physiological hypoxia” [16]. These low or hypoxic oxygen levels are sometimes beneficial for maintaining the functions of certain tissues. For example, hypoxia is necessary to maintain hematopoietic stem cell (HSC) homeostasis in the bone marrow [39]. Although HIF2α is dispensable, HIF1β is required for multiple HSC functions [4041]. The GC is also hypoxic due to the expansion of B cells. Moreover, hypoxia is known to affect the function of GC B cells [4243]. Female reproductive organs such as the vagina and placenta are hypoxic [4445], which is required for protection of the fetus from maternal immunity. For example, HIF1α induces the expression of trophoblast-regulating nonclassical class I histocompatibility antigens that prevent damage from natural killer (NK) cells [46].
ANTITUMOR CYCLE
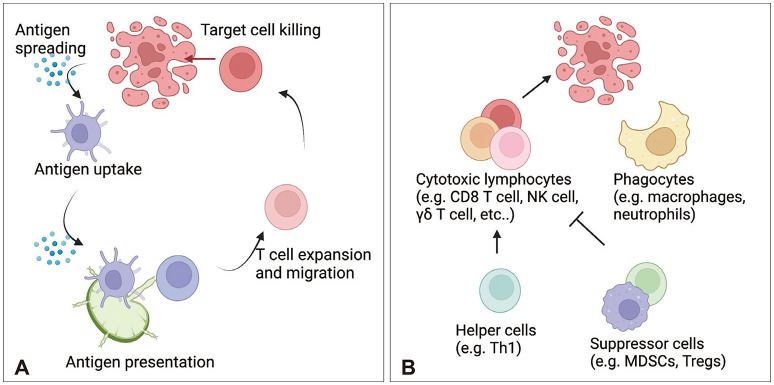
Multiple types of immune cells participate in antitumor responses. Following tumor cell death, antigen presenting cells (APCs), such as dendritic cells (DCs), mediate cellular immune responses by migrating to lymph nodes (LN) and presenting tumor antigens to T cells. Soluble antigens also drain into LNs and can be taken up by LN-resident DCs. CD8 T cells are considered the most important antitumor immune cell population. CD8a+/XCR1+/CD103+ DC1 cells can cross-present antigens to CD8 T cells via the major histocompatibility complex (MHC) class I, whereas CD4+/CD11b+/signal regulatory protein alpha (SIRPα)+ DC2 cells contain higher expression of MHC class II molecules and present antigens to CD4 T cells [47]. However, DC1 cells also prime CD4 T cells via MHC class II and CD40 [48]. Moreover, antigen-specific T cells can be primed by chemokines to migrate to the tumor site, recognize tumor cells via MHC molecules, and kill target cells. Tumor site APCs, such as macrophages, can stimulate the production of cytotoxicity and cytokine production by T cells or can prime them for exhaustion [49]. However, tumor cells can escape T cell responses via multiple mechanisms. For example, tumor cells downregulate MHC molecules and prime T cells for exhaustion via immune checkpoint molecules such as programmed cell death (PD) 1 and PD-1 ligand (PD-L1) [5051]. T cell responses can also be suppressed by anti-inflammatory cytokines such as IL-10 [52] and immune cells such as Tregs and MDSCs [53]. Therefore, researchers are investigating strategies to block inhibitory immune mediators. In addition to T cells, phagocytes including macrophages, microglia, and neutrophils can participate in the antitumor response via phagocytosis [545556]. Unconventional T cells such as NK cells, natural killer T (NKT) cells, mucosal-associated invariant T cells (MAIT cells), and γδ T cells are also involved in antitumor responses (Fig. 2) [5758].
ANTITUMOR RESPONSES IN THE BRAIN TUMORS
The brain is characterized as an “immune-privileged” organ. In 1921, a Japanese scientist attempted to transplant heterologous rat sarcoma tissue to the rat brain parenchyma [59], revealing that the tumor was not rejected. These observations were confirmed by James Murphy and Ernest Sturm using mouse sarcoma transplantation in the rat brain parenchyma [60]. Furthermore, the successful transplantation of skin autografts in the CNS has been demonstrated [61]. Based on these observations, the brain was thought to lack the ability to mount an immune response [62]. The idea of immune privilege was further supported by the notion that there is no lymphatic drainage from the CNS [6364]. However, the re-discovery of the dorsal and basal meningeal lymphatics led to the realization that immune system surveillance can occur around the CNS [656667]. For example, cerebrospinal fluid (CSF) produced from the choroid plexus can circulate through the CNS and the glymphatic system may allow an exchange between CSF and interstitial fluid (ISF). Due to the glymphatic exchange, parenchymal molecules can be drained by meningeal lymphatics [68]. CSF can reach the meninges, skull, and vertebral bone marrow [6669]. Additional drainage routes such as the cribriform plate also support drainage of brain-derived molecules [70]. As a result, antigen presentation primarily occurs at the dura and deep cervical lymph node (dcLN) [71]. Despite immune-surveillance by multiple cell populations, including DCs, immune tolerance is maintained within the CNS to prevent autoimmunity [72]. In addition, immune cell infiltration into the parenchyma of the CNS is restricted by multiple barriers including blood-brain barrier [73].
In addition to the barrier system, the brain tumor site is a unique microenvironment containing many immunosuppressive cells. Brain tumors are known as “cold tumors” due to the lack of neoantigens, low lymphocyte infiltration, and a predominant proportion of myeloid cells [74]. The nutrient-deprived TME is another factor contributing to immunosuppression [75]. Glucose deprivation inhibits immune cell activation and limits glycolysis. Due to the Warburg effect, lactate produced from glycolysis by tumor cells leads to enhanced Treg activation, decreased pH, and inhibition of T cell responses. As previously mentioned, brain tumors are also hypoxic [76], which can further induce T cell exhaustion, Treg cell migration, and γδ T cell malfunction [777879]. Thus, it is important to develop a comprehensive understanding of the complicated immunosuppressive brain TME to develop successful antitumor therapies.
THE ROLE OF HYPOXIA IN THE ANTI-BRAIN TUMOR IMMUNE RESPONSE
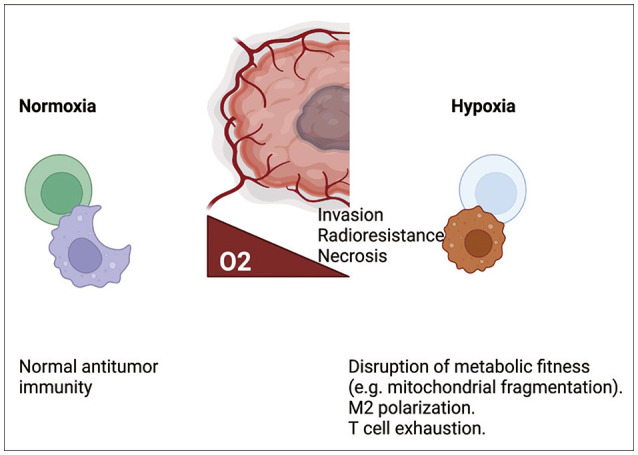
Hypoxia can affect immune cell responses through multiple pathways. Reductions in proliferation, cytokine production, and cytotoxicity, along with an increase in T cell exhaustion, have been observed in hypoxic CD8 T cell cultures [77] and in a murine melanoma model. In addition, exhausted CD8 T cells were more highly enriched in the hypoxic core regions of glioblastoma multiforme (GBM) tissue than in the peripheral regions [80]. Hypoxia also dampens the responses of T helper 1 (Th1) cells, important immune cells that help to mediate CD8 T cell responses and exhibit similar metabolic function, via HIF1α [81]. Several hypoxia-related pathways can promote T cell exhaustion, including PD-L1, a downstream HRE [82]. CD8 T cell exhaustion can also be induced via mitochondrial dysfunction produced by prolonged stimulation with hypoxia [83]; likewise, hypoxia attenuates NK cell responses via mitochondrial fragmentation [84]. A previous study reported that hypoxia is primarily driven by tumor cell oxygen consumption [85], and excessive tumor cell oxygen consumption reduces γδ T cell responses via hypoxia-mediated downregulation of NK group 2 member D (NKG2D) [7986]. Hypoxia can also facilitate the migration of Tregs [78], one of the various subsets of CD4 T cells that are metabolically adapted to hypoxic microenvironments via lactate-mediated stabilization [87]. In addition to T cells, hypoxia can affect macrophage immune responses by promoting M2 macrophage polarization in the glioma [88]. Thus, the effects of hypoxia on the immune system are mostly driven by the attenuation of antitumor immunity (Fig. 3). Therefore, targeting hypoxia may be an attractive option for antitumor therapy. We have shown that metformin, which is known to reduce mitochondrial respiration, reduced oxygen consumption in the glioma cell line GL261 and potentiated γδ T cell responses. Further, combination therapy with metformin or an HIF1α inhibitor administered concomitantly with γδ T cell therapy led to glioma transplant rejection in a murine model [79].
FUTURE DIRECTIONS FOR ANTI-BRAIN TUMOR THERAPIES
The primary therapy options for high-grade brain tumors include surgery, chemotherapy, and radiotherapy [89]. Despite standardized therapy, the average overall survival of patients is only 1–2 years [90]. Although immune checkpoint blockade has produced successful outcomes for certain tumor types, a recent clinical trial of anti-PD-1 had disappointing results [91]. Thus, further trials should be conducted. However, due to the unique immunosuppressive brain TME, it appears hard to be improved. Other studies have attempted to enhance antitumor responses via the abscopal effect using chemotherapy and radiotherapy [92]. However, a recent trial comparing radiotherapy combined with either nivolumab or chemotherapy did not show a beneficial effect with nivolumab [93]. In a murine brain tumor model, VEGF-C synergized with anti-PD-1 therapy to mediate the enhancement of meningeal lymphatics; therefore, it should be evaluated in human trials in the future [9495]. However, a combination of anti-cytotoxic T-lymphocyte associated protein 4 (CTLA4) and anti-PD-1 led to severe immune-related adverse effects (irAE) affecting multiple organs including the pituitary gland [96]. These adverse effects may be explained by the expression of CTLA4 within the pituitary gland [97]. In addition, disrupting immune tolerance within the healthy brain could trigger autoimmune diseases, which should be considered along with the risk for irAEs when developing future clinical trials. In addition to T cell-targeted therapies, targeting macrophages is also an attractive option. In previous studies, blocking M2 polarization via colony stimulating factor 1 (CSF1) and blocking the CD47-SIRPα-induced “don’t eat me” signal produced positive results in the murine model [9899]. However, CSF1 blockade showed disappointing results in another study [100].
Hypoxia induces abnormal vasculature, thereby limiting drug delivery [101]. Hence, acute hypoxia induced by Bevacizumab treatment may be one reason why it did not show favorable efficacy [102]. In addition, hypoxia facilitates M2 polarization of macrophages, limiting the efficacy of immune checkpoint inhibitors [8588], and induces radioresistance leading to dampened efficacy of radiotherapy [103]. Collectively, these findings support hypoxia as a suitable therapeutic target. If tumor cell-specific oxygen consumption can be reduced, we may be able to improve antitumor immune responses during immunotherapy. Reinvigorating the metabolism of tumor-infiltrating immune cells may be another option. A clinical trial evaluating the HIF2α inhibitor, PT2385, is currently ongoing. Other drug compounds, alone or in combination with immunotherapy, should also be evaluated in future trials [104].
CONCLUSION
Hypoxia is a hallmark of brain tumors that affects not only tumor cell characteristics, but also immune cells within the TME. Despite successful results with immunotherapies in multiple types of tumors, recent clinical trials for brain tumors have shown disappointing results. Thus, it is necessary to understand the complicated and unique characteristics of brain tumors. Because tumor cells exhibit high rates of proliferation, their metabolic demands are higher than other normal cells, thereby inducing nutrient deprivation in surrounding cells. The TME promotes hypoxia through the induction of abnormal vasculature and enhanced tumor cell oxygen consumption. Hypoxia may also be a critical factor contributing to the limited efficacy of antitumor therapy. Thus, understanding hypoxia in the brain TME is indispensable for improving strategies for developing antitumor drugs.
 XML Download
XML Download