

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hypophysitis (HP) is a rare disease which develops secondary to chronic or acute inflammation of the pituitary gland and may cause symptoms related to pituitary dysfunction and mass compression. HP can be classified based on the anatomical location of pituitary involvement (adenohypophysitis, infundibulo-neurohypophysitis, or panhypophysitis), the etiology (primary or secondary), and the histopathology (lymphocytic, granulomatous, xanthomatous, IgG4, necrotizing, or mixed forms) [1234]. Lymphocytic HP is the most common subtype of primary HP, while xanthomatous HP (XHP) is considered the rarest form, with 35 reported cases, to date [5].
The exact mechanism by which XHP occurs still remains unclear. The development of the characteristic histopathological lesion which consists of lipid-laden “foamy” histiocytes and CD68 positive macrophages has been previously attributed to the extension of an autoimmune/lymphocytic spectrum [6789]. On the other hand, various recent case presentations have reported to consider the inflammatory process of XHP related to the rupture of a Rathke’s cleft cyst (RCC) [4101112]. In our report, we describe a case with panhypopituitarism due to XHP secondary to the rupture of an RCC.
CASE REPORT
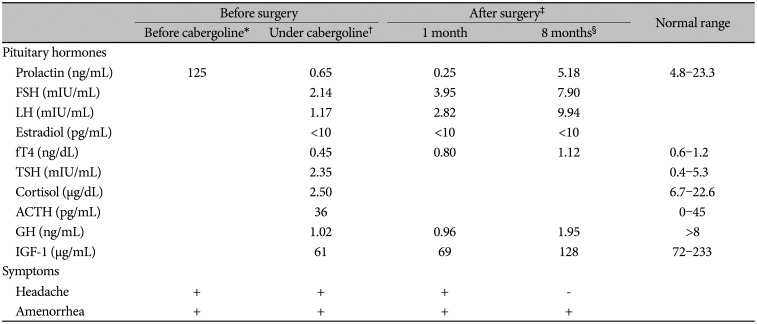
A 35-year-old woman was admitted to our Endocrinology clinic with a 2-year history of headache, which was bilateral frontal and episodic, and amenorrhea. She was initially admitted to a Gynecology clinic with amenorrhea 6 months prior to the admission to our institution. Initial laboratory testing revealed hyperprolactinemia with a prolactin level of 125 ng/mL (normal range: 4.8–23.3 ng/mL). Pituitary MRI showed a macroadenoma in the sella measuring 13×10×12 mm. Thus, she was started on cabergoline 0.5 mg twice a week; however, she was referred to an endocrinology clinic 6 months after starting the cabergoline treatment, due to persistent amenorrhea with low gonadotropins.
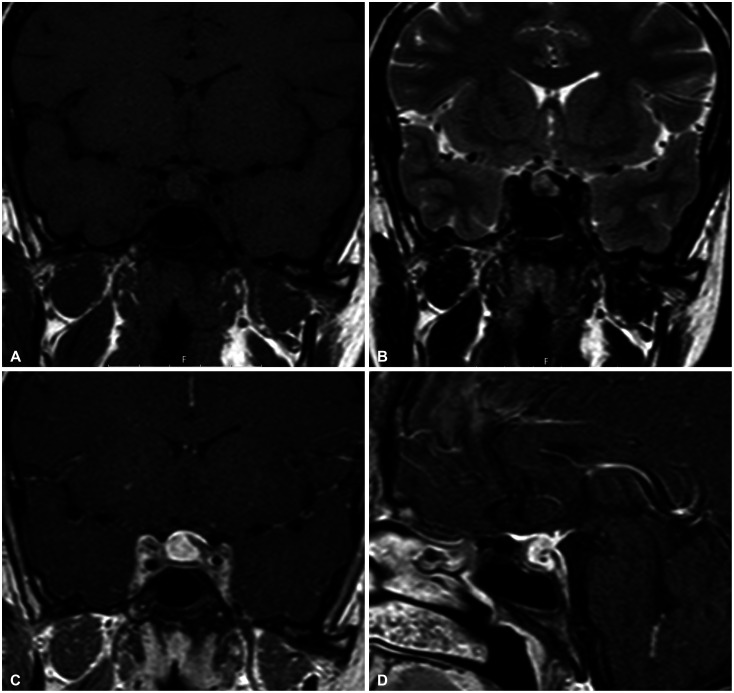
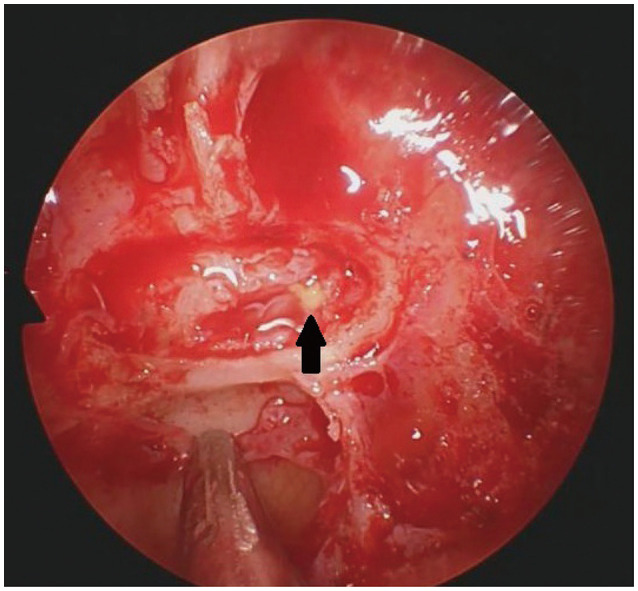
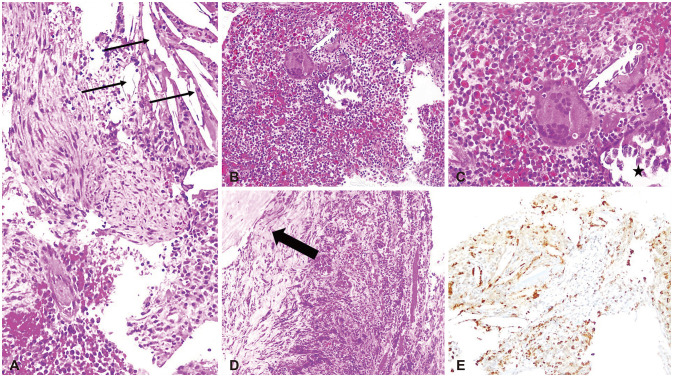
Her personal medical and family histories were unremarkable with no autoimmune disease. On physical examination, no abnormality observed in her temperature, blood pressure, body mass index, pulse and respiratory rates with a completely normal visual field. Her pituitary function profile revealed panhypopituitarism (Table 1) and a 13×11×12 mm sized sellar mass with diffuse enhancement which sustained toward the infundibulum and dura was observed on the gadolinium-enhanced pituitary MRI (Fig. 1). On both T1- and T2-weighted imaging, the lesion demonstrated heterogenous intensity. All these findings were reported as suggestive of a complicated cyst or cystic adenoma, hemorrhage, and/or hypophysitis. She was placed on hydrocortisone 10 mg twice daily, followed by a levothyroxine dose of 50 µg daily. As a diagnostic and therapeutic tool, the patient underwent transsphenoidal surgery for tumor resection with a steroid coverage. A standard endoscopic endonasal transsphenoidal approach was chosen. The opening in the sellar floor was made at the inferior side of the sella with a high-speed drill according to the location of tumor. After exposure of the dura, a small incision was made in the midline. Intraoperatively, thick yellowish fluid draining from the lesion was observed (Fig. 2). Cyst drainage was performed with an inverted T-shaped dural incision. When attempting to remove the cyst capsule, aggressive surgery was not performed in order to prevent damage to surrounding structures, such as the pituitary gland, pituitary stalk, and suprasellar cistern, and a portion of the cyst wall was allowed to remain. Histological examination revealed that the resected lesion was infiltrated with dominantly lymphoplasmacytic cells, foamy histiocytes, and only a small focus of cholesterol clefts and multinucleated giant cells. It also showed fibrosis whereas no granuloma or epithelioid cell was observed (Fig. 3). Immunohistochemically, these inflammatory cells were CD68, CD3, CD20, and CD138 positive. Immunostaining using pancytokeratin and epithelial membrane antigen immunostains were negative. Considering the preoperative imaging and the intraoperative macroscopic view, the final histopathological diagnosis was reported as a rupture of an RCC and an XHP. Tuberculosis and sarcoidosis were excluded by performing a tuberculin purified protein derivative test, a chest X-ray and measuring angiotensin converting enzyme level.
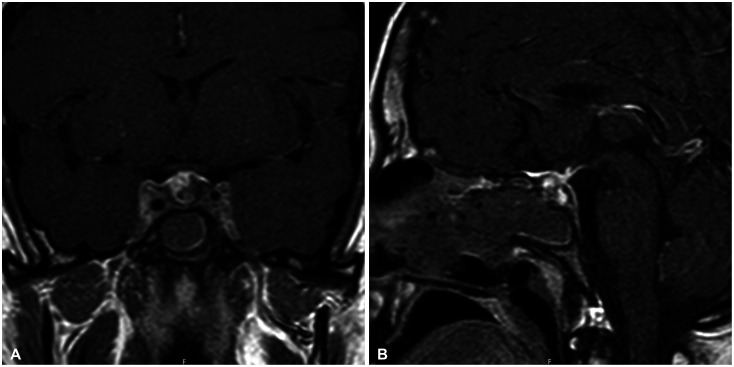
At 1-month follow-up postoperatively there was no improvement in the symptoms of our patient, i.e., amenorrhea, headache, and the lab results were still consistent with panhypopituitarism (Table 1). Gonadal hormone replacement therapy was not initiated in order to precisely observe the progression in the pituitary functions, and it was decided to follow the patient for 2 more months under the glucocorticoid and levothyroxine maintenance therapies. At the 3-month visit the patient was still complaining of headache and the absence of menstruation. In addition to that, a repeat pituitary MRI showed postoperative surgical changes with no remarkable residual lesion (Fig. 4). Consequently, the patient started high dose glucocorticoid therapy. A 64 mg daily dose of methylprednisolone was prescribed for 2 weeks and it was tapered by 4 mg weekly to the maintenance dose of 4 mg/day methylprednisolone. At 8-month follow-up postoperatively (4 months after methylprednisolone initiation), the patient stated that her headache completely resolved and a slight improvement was observed in the pituitary function tests (Table 1); however, there was no recovery in her menstrual cycle. Repeat pituitary MRI demonstrated no residual lesion. The patient still requires ongoing levothyroxine and glucocorticoid replacement and has not started gonadal hormone replacement therapy, yet, for the future evaluation of her gonadal axis. Gonadal hormone replacement therapy is planned to initiate 3 months later, if her menstrual cycle would not be recovered.
DISCUSSION
XHP is the rarest form of primary hypophysitis with a female preponderance (27/35 [77.1%]) and a mean age of 39.3 years [5]. The first three cases of XHP have been reported in 1998 [13], and since then, our 35-year-old female patient is the 36th reported XHP case worldwide and the second case in Turkey [14]. Headache, impaired menstrual cycles, hyperprolactinemia, diabetes insipidus, and panhypopituitarism have been reported as the most common presenting symptoms in a recent review [5] and our patient presented all these symptoms, except diabetes insipidus. In line with the literature which reported a low prevalence of visual disturbances in patients with XHP, no visual field abnormality was demonstrated in our patient [615].
Since the pathophysiological mechanism of XHP has not been elucidated, yet, XHP has been classified as primary hypophysitis [1]. A possible autoimmune mechanism which might be involved in the development of XHP has been suggested by three case reports, in which the cases with ulcerative colitis, Hashimoto’s thyroiditis, rheumatoid arthritis and Sjogren’s syndrome have been presented [789]. Dissimilar to these reports, recent evidences suggest that the inflammatory process in a xanthomatous lesion may actually be a secondary response to mucous fluid content release from a ruptured cyst, most commonly from an RCC [51216171819202122]. Moreover, Duan et al. [11] have presented a cohort of 7 XHP cases, in which 6 cases harbored concurrent findings of a ruptured RCC and the last case had a history of a surgery for an RCC. In the same cohort, thick yellowish drainage within a cystic lesion was observed in 4 cases intraoperatively, which was similar to that described in our case. The histopathological evaluation of our case also showed a focal area of cholesterol clefts and multinucleated giant cells which might support the impression that there was an overlap between XHP and xanthogranulomous hypophysitis [12].
The main response of a mammalian tissue to injury is inflammation and this damaged tissue tends to be replaced by parenchymal/connective tissue elements or fibrosis. The association between hypophysitis and RCC has been attributed to the inflammatory reaction induced by the cyst wall or the leakage of the cyst contents [2324]. The inflammatory response of tissue in the epithelium may be triggered by the secreted mucus, a strong stimulator of tissue, from goblet cells in the cyst wall. In accordance with various case reports [24], despite the absence of a direct evidence for a cyst rupture histopathologically, the possible etiology for XHP in our case was considered as a ruptured RCC, according to the intraoperative evaluation of the lesion by the experienced neurosurgeons at our institution.
Gadolinium-enhanced pituitary MRI is the preferred modality for hypophysitis, and XHP usually presents as a cystic sellar mass, which generally shows a peripheral enhancement [2]. In addition to that, Mathkour et al. [5] reported a case, in which the sellar lesion had a suprasellar expansion with the infiltration of the cavernous sinus which has been considered as adenoma over RCC on MRI. As a result, it may be difficult to differentiate XHP on MRI from a cystic adenoma, RCC, hemorrhage, or a pituitary abscess. Heterogenous intensity on both T1- and T2-weighted imaging and diffuse pituitary enhancement which was displayed by MRI in our case were primarily suggestive of a complicated cyst/cystic adenoma or hemorrhage, while the infundibular and dural enhancement indicated a diagnosis of hypophysitis, as well.
The management of XHP may be challenging due to the ambiguous pathogenesis of the disease. Unlike the treatment for lymphocytic hypophysitis, glucocorticoid therapy is less effective in XHP and a surgical intervention is often required to alleviate the compressive symptoms and pituitary dysfunction [46]. An XHP case which was radiologically responsive to high dose steroid has been described by Joung et al. [9] and the authors have reported a significant mass reduction at the sella and suprasellar area following a 500 mg intravenous methylprednisolone treatment for 3 days. Similarly, in a recent report which presented a 14-year-old prepubertal girl with XHP, a reduction in the mass size as a response to high dose prednisolone treatment for 6 months was demonstrated. However, the authors reported that the effect of steroid therapy was not sustained, thus azathioprine was added as a steroid-sparing agent, which maintained the reduced mass size [25]. In addition to playing an essential role in making the histological diagnosis of XHP, surgery has been generally considered in cases with progressive visual field deficits or impaired pituitary function [6]. In the literature, the first case with XHP which had a full recovery of pituitary dysfunction following a transsphenoidal surgery has been reported by Burt et al. [16] and that patient had only a 3-month history of hypogonadism at the initial admission. In another case report, diabetes insipidus completely cured after surgery in an XHP patient with a 4-month history of polyuria and polydipsia [26]. Similarly, a case of XHP with a 2-month history of amenorrhea, galactorrhea, and mild weight gain achieved remission following a complete resection of the lesion [5]. The rate of pituitary function improvement following surgery in XHP cases has been reported less than 50%, which attributed to the duration of the impairment [8]. Chronic inflammation has been considered responsible for irreversible pituitary dysfunction via fibrosis and tissue destruction. In addition to that, headache has been reported to improve in most of patients with XHP who had surgery [15]. Headache and panhypopituitarism of our patient did not recover within 6 months after surgery, possibly due to a 2-year history of the presented symptoms. Interestingly, headache disappeared completely within a few days after the first dose of high dose steroid therapy. Moreover, the pituitary function tests showed a slight improvement, i.e., normalization of IGF-1 level and a mild increase in the other anterior pituitary function tests; however, at 10-month follow-up, our patient has still been requiring hormone replacement therapies.
XHP is an uncommon inflammatory process of the pituitary gland which should be included in the differential diagnoses, if a patient presents with headache, panhypopituitarism and if yellowish fluid is drained from a cystic lesion intraoperatively. In agreement with the recent reports, our case presentation may also indicate a recommendation to classify XHP as secondary hypophysitis which could be caused by a ruptured cystic lesion. Here, we presented a patient with XHP which developed as an inflammatory response to a ruptured RCC and the transsphenoidal surgery did not improve the symptoms/pituitary functions, while headache recovered immediately after the first dose of high dose methylprednisolone treatment.
 XML Download
XML Download