

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Optic pathway gliomas (OPGs) are insidious, debilitating low-grade gliomas (LGGs) that account for 3%–5% of all pediatric brain tumors. Histologically, OPGs are primarily pilocytic astrocytomas (World Health Organization grade I), although pilomyxoid astrocytomas and grade II diffuse fibrillary astrocytomas have also been reported [12]. They are common in patients with neurofibromatosis type 1 (NF1), with up to 20% developing OPG at a mean age of 4.5–5.0 years [3]. NF1 is caused by pathogenic variants of the NF1 gene, located on chromosome 17q11.2. The NF1 gene encodes the neurofibromin protein, a tumor suppressor. Conversely, children without NF1 may develop sporadic OPG [4]. The molecular pathogenesis of sporadic OPGs is yet to be understood, but the most common genetic alteration in sporadic tumors is BRAF duplication [56].
OPGs most commonly arise in the chiasmatic-hypothalamic region but can arise anywhere along the optic pathway. Depending on their location, OPGs can result in a variety of symptoms and signs, including decreased visual acuity (VA), proptosis, strabismus, nystagmus, headaches, seizures, and precocious puberty. The location of OPGs typically precludes complete surgical resection or optimum radiation dosing without incurring an often-unaccepted neurological sequelae. The complexity of symptomatology and its close relationship to key structures make the treatment of OPG challenging. Treatment decisions should consider the patient’s age, presence or absence of NF1, and location of the tumor. Therefore, OPGs require multidisciplinary care by neurosurgeons, oncologist, radiation oncologists, endocrinologists, ophthalmologists, pathologists, geneticists, and health-care professionals, and their management should be highly individualized. OPG growth alone may not be an indication for treatment, depending on the patient’s current vision. Treatment is usually indicated in the case of radiological or clinical progression, such as significant visual deterioration of neurological symptoms. Because, OPG may remain stable in volume (presumed to be mostly NF1) or, rarely, regress spontaneously in the case of NF1 associated OPG [7]. The unique biology of these lesions results in numerous recurrences or progressions in many patients, necessitating additional therapies and consequent cumulative toxicities [8].
Currently, the focus of ongoing studies on OPG is related to pharmacological agents. This review includes standard and emerging therapies for OPGs, with a summary of the roles of chemotherapy and molecularly targeted therapies.
STANDARD CHEMOTHERAPY
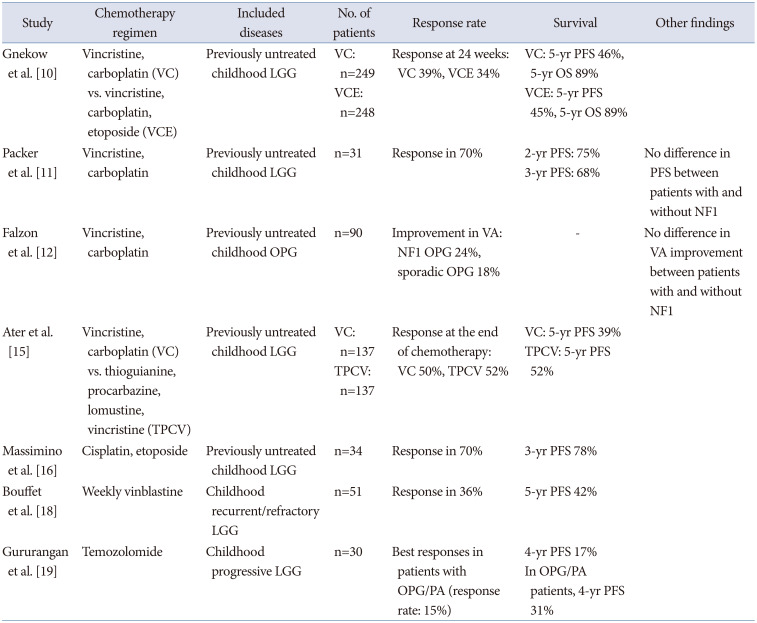
Despite the benign histological appearance of many OPGs, chemotherapy results in high response rate. For this reason, chemotherapy is often preferred as an initial treatment because surgery is often limited or not feasible owing to the risk of damaging visual, neurological, or endocrine function [9]. Chemotherapy can often postpone the need for radiation, a delay that may reduce neurocognitive morbidity without compromising survival. Between 40% and 60% of patients progress during or after first-line chemotherapy and successive systemic treatments are often necessary. Patients with OPGs in the setting of NF1 often have indolent disease, and fewer patients require subsequent therapy. Sporadic OPGs confer a significantly higher risk of vision loss than those secondary to NF1.
Carboplatin-based chemotherapy is often the first-line treatment for patients with progressive disease. In Europe, treatment with carboplatin and vincristine over an 18-month period represents the current first-line strategy in the Societe Internationale d’Oncologie Pediatrique (SIOP), despite modest visual outcomes [10]. For patients with NF1 who received carboplatin and vincristine, the 3- and 5-year progression-free survival (PFS) rates were 77% and 69%, respectively [11]. In a prospective study comparing post-chemotherapy VA outcomes in patients with OPG with and without NF1, there was no difference between the groups, with 24% improved, 35% remained stable, 41% worsened in the NF1 group and 18% improved, 43% remained stable, and 39% worsened in the sporadic group [12]. This result is similar to those of previous studies [13]. It should be noted that there is a poor correlation between radiographic and VA results in numerous studies [1314]. Hence, it is essential to focus on the clinical effects of therapy on visual function.
Alternative therapies such as the thioguanine, procarbazine, lomustine, vincristine (TPCV) showed a nonsignificant trend toward improved event-free survival when compared with carboplatin/vincristine in patients with NF1 [15]. The combination of cisplatin and etoposide has also been evaluated in the treatment of OPGs, with a 3-year PFS rate of up to 78% [1617]. However, this regimen should be used cautiously because of the risk of secondary leukemia associated with etoposide and ototoxicity associated with cisplatin. Vinblastine monotherapy is a commonly accepted chemotherapy for OPG. In a phase II study of vinblastine monotherapy for children with recurrent pediatric LGGs, there was 36% response rate, including “minor responses” which was defined as shrinkage between 25% and 49% [18]. The 5-year PFS rate in this study was 42.3% [18]. In recent years, monotherapies with temozolomide and vinorelbine have also been used for progressive or refractory disease with positive results and low toxicity. In a phase II study of temozolomide in children with progressive OPG and pilocytic astrocytoma, the best responses were partial response (PR) in 11%, minor response in 4%, stable in 38% [19]. There have been reports of secondary acute leukemias after a short latency period following temozolomide in adult patients who received temozolomide concurrently with radiotherapy [20]. Concerns could be raised regarding the long-term complications of using a DNA-methylating agent in patients with an inherent genetic predisposition to tumors, especially leukemia. A summary of clinical data of chemotherapies for OPGs is present in Table 1.
ANTI-VASCULAR ENDOTHELIAL GROWTH FACTOR AGENT
The anti-vascular endothelial growth factor (VEGF) agent bevacizumab was introduced in 2009 as the next line treatment for progressive OPG, as angiogenesis plays an important role in the growth of LGG [21]. Increased microvascular density in OPGs has been associated with worse PFS [22]. VEGF induces neovascularization and is abnormally expressed in glial neoplasm [23]. By inhibiting VEGF, tumor growth and vascular permeability are reduced.
Treatment outcomes show a rapid radiological response with anecdotally profound visual improvement [2425]. Bevacizumab-based therapy has achieved objective responses and rapid improvement in visual symptoms in up to 86% of refractory cases [26]. Combination therapy with bevacizumab and irinotecan achieved a 2-year survival rate of 47.8% in patients with recurrent LGGs [27]. Bevacizumab monotherapy did not appear to decrease the efficacy of treatment and reduced toxicity when compared with combination therapy [26]. According to Hwang et al. [26], approximately 86% of patients achieved an objective response with a median time to maximal response of 9 weeks. Bevacizumab monotherapy is also effective in improving visual deterioration without tumor progression and the visual field improved by bevacizumab could be independent of imaging changes [28].
Recurrence or progression after discontinuation of bevacizumab is frequent, with a relapse rate of 15%–83% within 6 months after cessation [2529]. However, retreatment with bevacizumab after relapse can achieve good responses [2530]. An ongoing study (NCT02840409) enrolled newly diagnosed patients with LGGs, including those with NF1, treated with vinblastine alone or vinblastine plus bevacizumab. The hypotheses underlying this approach are that bevacizumab will result in a greater occurrence of visual and neurological improvement, whereas vinblastine will result in longer disease control after 6 months of treatment compared with the historical experience with bevacizumab alone.
The most common side effects of bevacizumab include hypertension, fatigue, joint pain, bleeding, and proteinuria. However, these effects are usually reversible after the discontinuation of bevacizumab. Given the good visual outcomes of bevacizumab-based therapy, it could be an option for patients with refractory diseases. The optimal duration of therapy has not been defined; however, it seems plausible to consider longer courses of therapy if tolerated.
MITOGEN-ACTIVATED PROTEIN KINASE PATHWAY INHIBITOR
Recently, the effect of targeted inhibition of mitogen-activated protein kinase (MAPK) pathways on LGG is being increasingly studied in terms of dose, treatment duration, effectiveness, and toxicity. It is now well understood that abnormal activation of MAPK signaling pathways, such as Ras and Raf is the most frequent genetic aberration observed in progressive LGGs, most commonly resulting from activation of the BRAF oncogene [3132]. The two most common aberrations of BRAF are tandem duplication, resulting in KIAA1549-BRAF fusion, and an activating point mutation, BRAFV600E. Regarding other gene mutations, gene mutations and fusion gene formation in FGFR1 and fusion gene formation in the NTRK family have been reported. These abnormalities activate the MAPK/ERK signaling pathway [33].
In cases of NF1, the NF1 gene controls the RAF-MEK-ERK signaling pathway downstream by controlling RAS, thus activating ERK, which is a typical signaling pathway of the MAPK during NF1 gene mutation. Alterations in the MAPK pathway including interactions with aberrations common to NF1, lead to propagation of LGG.
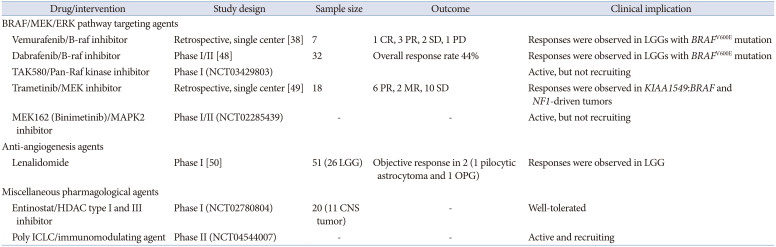
Numerous novel agents, especially MEK inhibitors, are currently in clinical trials targeting the MAPK pathway. MEK inhibitors such as selumetinib, refametinib, trametinib, and cobimetinib have recently been used in the treatment of progressive and recurrent LGGs in children, with a 20-year PFS rate of up to 69% [34]. These agents target a downstream mediator in the Ras signaling pathway, preventing constitutive MAPK activation. Their efficacy is likely to be greatest in patients with BRAF mutations [34]. As a MEK1/2 inhibitor, selumetinib avoids the adverse event of paradoxical activation of the MAPK pathway that occurs when BRAF-KIAA1549 aberrant pediatric LGGs are treated with direct BRAF inhibitors [35]. In a phase II trial of selumetinib in children with recurrent OPGs without NF1, the 2-year PFS rate was 78% [36]. Imaging responses were PR in 24%, stable in 56%, and progression in 20%. In terms of visual outcomes, 21% and 68% of patients improved and were stable, respectively. Another phase II trial of selumetinib in pediatric patients with BRAF-aberrant or NF1-associated recurrent LGGs reported a 40% response rate, stable-to-improved VA in patients with OPG, and a 2-year PFS rate of 96% [37]. Selumetinib is an oral agent that requires fewer clinic visits. According to the Pediatric Brain Tumor Consortium (PBTC) phase II trial of selumetinib, patients were seen monthly [36]. Common toxicities include creatinine phosphokinase elevation, anemia, diarrhea, headache, fatigue, and rash, which are relatively tolerable. Considering manageable toxicities, fewer clinic visits, and the absence of a central line/intravenous access would favorably affect a patient’s quality of life compared to standard chemotherapy.
Selective type 1 B-Raf competitive small-molecule enzyme inhibitors including vemurafenib and dabrafenib recognize and bind to the ATP-binding domain of BRAFV600E-mutants. This interrupts the B-Raf/MEK step in the B-Raf/MEK/ERK pathway, which drives tumorigenesis in BRAFV600E-mutant LGGs. Vemurafenib has shown some promise for the treatment of BRAFV600E-mutant LGGs [38]. Of the seven patients who were treated with vemurafenib for BRAFV600E-mutated LGGs, the best responses to treatment were as follows: 1 complete response (CR), 3 PR, 1 stable and 1 progression, respectively. Treatment was well tolerated, with dermatological toxicity being the main concern [38].
MAMMALIAN TARGET OF RAPAMYCIN INHIBITOR
The mammalian target of rapamycin (mTOR) serves as a pivotal signaling pathway that regulates key cellular processes, including metabolism, protein synthesis, cell cycle progression, angiogenesis, and apoptosis [39]. Both NF1-associated and sporadic LGGs have demonstrated abnormal signaling upstream of mTOR through mutations in receptor tyrosine kinase, or more commonly in sporadic LGG, through alterations in BRAF [540]. Everolimus is a macrolide derivative of rapamycin that selectively inhibits mTOR. It can be orally administered and has been used extensively in both adults and children, including prolonged use in organ transplant recipients and children with subependymal giant cell astrocytoma [41]. Given the well-tolerated toxicity profile of everolimus and the central role of the Ras/Raf/mTOR pathway in pediatric LGGs, everolimus is being investigated for the treatment of LGGs.
In a phase II study of everolimus in 23 recurrent and/or progressive pediatric LGGs including six OPGs, responses to treatment by week 48 were as follows: 2 PR, 10 stable, and 11 progression, respectively [42]. In a recently published Neurofibromatosis Clinical Trials Consortium phase II trial of everolimus, 23 patients with LGGs were enrolled, including 13 with OPGs. Fifteen (68%) patients demonstrated a response (1 CR, 2 PR, and 12 stable) [43]. The favorable toxicity profile observed in NF1 populations did not significantly differ from that in other populations [44]. Regarding VA, Ullrich et al. [45] reported that the majority of children with NF1-OPG exhibited stabilization of their VA after everolimus treatment with 4/25 eyes improved, 19/25 eyes stable, and 2/25 eyes worsened.
Currently, many new agents for LGG target the BRAF/MEK/ERK pathway, RAS pathways, angiogenesis, immunomodulation, and the tumor microenvironment. A summary of other new agents under investigation is present in Table 2.
FURTHER CONSIDERATION FOR TREATMENT
Several important issues in treating OPGs remain uncertain. One of the most important considerations is regarding the natural history of OPG and prediction of outcome. Tools to predict the clinical course and long-term outcomes of patients with OPGs are lacking. It is controversial which outcome measures should be used. Commonly used oncological outcome measures, such as overall survival and radiological PFS, may not be the most appropriate for evaluating OPGs. Survival rate and tumor shrinkage are not as important as functional outcomes, especially given the excellent overall survival children with OPGs. Visual function, endocrine/hypothalamic dysfunction, and quality of life measures should be considered when measuring the outcomes. In the same context, newer drugs, including molecular targeting agents, should be used earlier in the disease process, compared with standard chemotherapies, and be assessed for efficacy not only for radiological response and survival, but also for functional outcomes. Furthermore, the optimal timing of therapy initiation and its influence on the overall outcomes are not fully known. It will also be important to not only focus on functional outcomes but also better understand the length of therapy required, duration of response/stability, and late effects of treatment [4647].
CONCLUSION
The management of OPGs remains challenging and our understanding of their behavior continues to evolve. Treatment is usually unnecessary for OPGs that do not cause visual impairment. When patients manifest a decline in VA, visual field or significant radiological progression, chemotherapy with vincristine and carboplatin remains the first-line treatment. Newer molecular targeting agents such as anti-VEGF agents, MEK inhibitors, and mTOR inhibitors, have shown promising outcomes in relapsed and progressive cases. With improvements in molecularly targeted therapies, the long-term impact and visual morbidity will be reduced for patients with OPG.
 XML Download
XML Download