

 PDF
PDF Citation
Citation Print
Print


Introduction
Periodontitis is a chronic inflammation that occurs in deep periodontal tissues and can lead to attachment loss, alveolar bone resorption, and eventually tooth loss (1, 2). Bone regeneration is the main objective of periodontal therapy. Tissue engineering has recently provided alternative technologies to regenerate periodontal bone tissues by transplanting appropriate cells and scaffolds (3, 4). Stem cells from human exfoliated deciduous teeth (SHED) are a type of mesenchymal stem cells with strong osteogenic and proangiogenic properties (5-7). When combined with various scaffolds, SHED have the potential to enhance bone defect repair (8-10). However, little is known about the cellular and molecular mechanisms underlying SHED promotion of efficient bone repair. The question of whether a direct action through differentiation of the implanted cells or a paracrine role through growth factor secretion remains unresolved (10, 11).
Cell tracking in vivo is an important method to study the molecular mechanisms by which transplanted cells promote bone regeneration. In this study, magnetite (Fe2O3)-based superparamagnetic iron oxide nanoparticles (SPIOs) were used as the labeling agents for magnetic resonance imaging (MRI) cell tracking. Transplanted areas containing SPIO-labeled cells will appear as regions of low signal intensity, creating negative contrast on T2 weighted MRI (12). However, previous research has suggested that SPIO particles may be phagocytized by macrophages, resulting in the division and death of labeled cells as well as the formation of false-positive MRI results (13). Fluo-rescence-labeled SPIO, such as Molday ION Rhodamine-BTM (MIRB), can easily be detected and tracked by fluorescence microscopy and confirmed by MRI scans, making it a superior SPIO-labeled material (14). This technology not only allows real-time identification of transplanted stem cells using MRI but also allows detection of transplanted cells using histochemistry (15). A high dose of MIRB is toxic to cells and inhibits cell proliferation and differentiation; however, a low MIRB dose will influence the MRI tracer signal. Consequently, we must establish an optimal labeling concentration of MIRB by analyzing the proliferation and differentiation of SHED labeled with various MIRB concentrations (16).
MIRB concentration was set at 25 μg Fe/ml in this investigation because it not only satisfied the signal intensity of MRI detection but also did not have a negative impact on cell proliferation and differentiation. MRI, HE staining, and immunohistochemistry results indicated that both labeled and unlabeled SHED transplanted to the periodontal bone defect region could promote bone rege-neration. The colocalization of MIRB and hNUC showed that the transplanted SHED could survive in vivo. Immu-nofluorescence and enzyme-linked immunosorbent assay (ELISA) revealed that SHED could not only differentiate into osteoblast-like and osteocyte-like cells but it could also secrete osteogenic proteins to mobilize host cells. These findings suggested that SHED might stimulate bone repair through both direct cell differentiation and indirect paracrine processes.
Go to : 
Materials and Methods
Cell culture and osteoinductive treatment
Deciduous teeth were extracted from healthy children aged 6∼8 years following a procedure approved by the Ethics Committee of Liaocheng People’s Hospital (Liaocheng, China). SHED were isolated from the dental pulp using an outgrowth method as described in a previous report (17). For osteoinductive treatment, cells were seeded at a density of 2×105 cells/well in a six-well plate and cultured in an osteoinductive medium for 2 weeks. The medium was replaced twice a week. The osteoinductive medium was composed of the following ingredients: 90% DMEM (Gibco, Carlsbad, CA, USA), 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 100 U/ml penicillin (Gibco, Carlsbad, CA, USA), 100 U/ml streptomycin (Gibco, Carlsbad, CA, USA), 100 nmol/l dexamethasone, 10 mM β‑glycerol phosphate, 50 nM vitamin D3, and 50 μM l-ascorbic acid 2-phosphate (Sigma‑Aldrich, St. Louis, Missouri, USA). Alizarin Red S staining was used to detect the mineral deposit 2 weeks after osteogenic induc-tion. Cells were fixed with 4% paraformaldehyde (m/v; Sigma-Aldrich, St. Louis, Missouri, USA) and then stai-ned with 40 mM Alizarin Red S (pH 4.2; Sigma-Aldrich, St. Louis, Missouri, USA) for 10 min at room temperature. Cells were observed, and images were captured with an inverted fluorescence microscope (Nikon Ti, Tokyo, Japan).
Western blot analysis
Total protein was isolated from the osteogenic induction group and non-treated group with RIPA buffer and quantified with a BCA protein assay kit (Beyotime, Beijing, China). Equal quantities of samples were separated via 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a hydrophi-lic polyvinylidene fluoride (PVDF) membrane (0.45 μm, Merck Millipore, Boston, MA, USA) with the Bio-Rad protein assay system (Bio-Rad, Hercules, CA, USA). Primary Antibodies against Runx2 (1:1,000, Abcam, Cambridge, UK), Alp (1:1,000, Abcam, Cambridge, UK), and β-ac-tin (1:2,000, Santa Cruz Biotechnology, CA, USA) were incubated with the membrane at 4℃ overnight. The secondary antibodies, horseradish peroxidase (HRP)-linked goat anti-rabbit IgG or goat anti-mouse IgG (Santa Cruz Biotechnology, CA, USA), were incubated with the membrane at room temperature for 1 h. The membrane was immersed with electrochemiluminescence (ECL) response solution (Pierce, Waltham, MA, USA) at room temperature for 1 min. All bands were analyzed with Image Lab Software (version 5.1; Bio-Rad Laboratories, CA, USA), and relative levels against the internal control β-actin were calculated.
Fluorescence observation of MIRB-labeled SHED
Cells were seeded in six-well plates at a density of 2×105 cells/well. MIRB (BioPAL Co., Worcester, MA, USA) stock solution (2 mg Fe/ml) was added to cell culture medium (DMEM+10% FBS) to prepare labeling medium with final concentrations of 0, 12.5, 25, 50, and 100 μg Fe/ml. SHED were then incubated in the labeling medium for 24 h under standard culture conditions (37℃, 5% CO2, humidified). Cells were washed three times with PBS and fixed with 4% paraformaldehyde for 15 min. After washing the cells three times with PBS, these were incubated with a blocking solution (5% BSA+1% Triton X-100 in PBS) for 1 h. A FITC-phalloidin conjugate solution (Enzo, New York, NY, USA) was then used to stain filamentous actin at room temperature for 1 h. The cells were again washed three times with PBS and then observed under a fluorescence microscope.
Prussian blue staining and labeling efficiency calculation
SHED were labeled with various MIRB concentrations, and then fixed with 4% paraformaldehyde. SHED were washed three times with PBS and then stained with Perl’s Prussian blue reagent (2% potassium ferrocyanide in 6% hydrochloric acid) (Leagene Biotechnology, Beijing, China) for 30 min. The intracellular distribution of Fe3+ was observed under a light microscope. In general, we selected 10 fields of view randomly to count Prussian blue positive stained cells under a 10-fold microscope. The labeling efficiency was determined using the following equation: Labeling efficiency (%)=(number of Prussian blue positive cells/number of total cells)×100%.
Detection of intracellular iron content
The average intracellular iron concentration was determined using an iron assay kit (Biovision, Inc., CA, USA). Cells were plated in six-well plates at a density of 2×105 cells/well and incubated with a MIRB labeling medium for 24 h. Labeled SHED were then lysed with 65 μl of Iron Assay Buffer. The lysate was centrifuged at 16,000×g for 10 min. A 50 μl volume of supernatant was added to each well of a 96-well plate, and an equivalent amount of Iron Assay Buffer was added to create a total volume of 100 μl. A 5 μl iron reducer was added to each well to convert Fe3+ to Fe2+. A 100 μl of iron probe was added to each well and incubated for 60 min at room temperature. The absorbance was read with a spectrometer at 593 nm. Mean while, an iron content standard curve was created according to the kit instruction. The total iron content in each well was determined according to the standard curve. The number of cells in each well was counted with a hemocytometer. The iron content was determined using the following formula: The iron content of a single cell=Total iron content per well/Number of cells per well.
Trypan blue staining and viable cell percentage calculation
We prepared a 4% Trypan blue stock reagent by dissolving 4 g of Trypan blue (Merck KGaA, Darmstadt, Hessen, Germany) in 100 ml of double-distilled water and filtering the solution through a filter paper. The stock reagent was diluted to 0.4% with PBS before use. A single-cell suspension was prepared from five groups of cells labeled with 0, 12, 25, 50, and 100 μg Fe/ml MIRB, respectively. An equal amount of 0.4% Trypan blue and single-cell suspension were mixed, and the mixture was incubated for approximately 3 minutes at room temperature. Then, live and dead cells were counted individually under a microscope (dead cells were stained light blue and live cells were colorless). The percentage of viable cells was computed using the following formula: Live cells percent (%)=Total number of live cells/(Total number of live cells+Total number of dead cells)×100%.
Effect of MIRB labeling on the proliferation of SHED
SHED were seeded in a 96-well plate at a density of 1,000 cells/well. Once the SHED adhered to the wall of the wells, they were incubated with a labeling medium containing various concentrations of MIRB (0, 12.5, 25, 50, and 100 μg Fe/ml) for 24 h. After that, the labeling medium was replaced with 100 μl of a standard culture medium and the SHED were cultured for an additional 1, 3, 5, and 7 d. At each time point, 10 μl of CCK-8 solution (Yeasen, Shanghai, China) was added to each well of the plate and the mixture was incubated at 37℃ for 2 h. Finally, absorbance at 450 nm was determined using a microplate spectrophotometer (Bio-Rad, CA, USA).
Effects of MIRB labeling on the osteogenic differentiation of SHED
SHED were seeded in a six-well plate at a density of 2×105 cells/well. The cells were incubated with a labeling medium containing different concentrations of MIRB (0, 12.5, 25, and 50 μg Fe/ml) for 24 h. Labeled SHED were then cultured in an osteoinductive medium for 2 weeks. The cells were fixed with 4% paraformaldehyde and stained with 2 ml of Alizarin Red S solution (pH 4.2; Sigma‑Aldrich, St. Louis, Missouri, USA) for 15 min. Images were captured using an inverted fluorescence microscope. For semi-quantitative analysis of Alizarin Red S, 600 μl of cetylpyridinium chloride solution (100 g/l; Sigma‑Aldrich, St. Louis, Missouri, USA) was added to each well and incubated for 15 min at room temperature. Absorbance was read using a spectrometer at 562 nm (Bio-Rad, CA, USA).
MRI analysis of MIRB labeled SHED in vitro
SHED labeled with different concentrations of MIRB (0, 12.5, 25, and 50 μg Fe/ml) were collected and counted with a hemocytometer. One million cells from each group were transferred to1.5-ml centrifuge tubes (Eppendorf, Westbury, NY, USA). After centrifugation at 150×g for 5 min, SHED were resuspended in 15 μl of thrombin solution. Then, the 15 μl of thrombin solution containing 1×106 cells and 15 μl of fibrinogen solution were simultaneously injected into the bottom of the 1.5-ml centrifuge tube. The mixture condensed to jelly in 1∼2 min. The tubes were then imaged on a 1.5-T system (Achieva, Philips Healthcare, Best, The Netherlands) with an eight-channel wrist coil (repetition time [TR]=20 ms, International time [TE]=8.1 ms, flip angle=25, field of view [FOV]=80×80×30 mm3, slice thickness=4 mm, and Mat=208×208).
Animals and group design
All animal experiments were performed following a protocol approved by the Animal Committee of Liaocheng People’s Hospital (Liaocheng, China). Forty-eight SD rats (6∼8 weeks old, 200∼250 g, male) were numbered and divided randomly into four groups: (1) SHED (MIRB), (2) SHED, (3) Fibrin, and (4) PBS. Each rat has a defect in the right mandible. Six rats from each group were anesthetized at 0, 3, 6, and 9 weeks after the surgery. The healing of the defect was captured by MRI. At 2 and 4 weeks after the surgery, three rats in each group were perfused to obtain samples of periodontal bone defects for histological and immunohistochemical analyses.
Preparation of periodontal bone defects in rats
Periodontal bone defects were constructed according to the following procedures. The rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (4 ml/kg). An approximately 2 cm long extraoral incision was made parallel to the lower border of the rats’ mandibles. The subcutaneous tissues and masseter muscles were separated from the alveolar bone surface. A periodontal wound defect (length×height×depth: 5 mm×4 mm×1 mm) was constructed with a low-speed dental round bur, accompanied by physiological saline irrigation. The defect was located at 1 mm behind the anterior mandible and 1 mm below the crest of the alveolar bone. The muscle and skin were sutured, and the wound was cleaned with iodophor after surgery. To prevent the immune system from rejecting the transplanted cells, we intraperitoneally injected cyclosporine A (10 mg/kg; Sigma-Aldrich, St. Louis, Missouri, USA) into the rats 24 h before transplantation and every day after the surgery until the animals were euthanized.
Preparation of fibrin glue containing SHED for transplantation
Fibrin glue was prepared with a fibrin sealant kit (SHANGHAI RASS, Shanghai, China). One million fresh SHED (MIRB-labeled or unlabeled) were resuspended in a 15 μl thrombin solution (450 IU/ml) for cell trans-plantation. The thrombin solution containing SHED (labeled or unlabeled) was injected into the defect sites of rats. Simultaneously, an equal amount of fibrinogen solution (40 mg/ml) was injected into the same site. In the rats in the Fibrin group, both thrombin and fibrinogen solutions were injected into the defect site, whereas in the rats in the PBS group, only PBS was injected.
MRI evaluation and analysis after transplantation of SHED into rats
MRI analysis on rats was performed on a 1.5-T system (Achieva, Philips Healthcare, Best, The Netherlands) with an eight-channel wrist coil at 0, 3, 6, and 9 weeks after the surgery. Subjects were positioned face down in the prone position. Three-dimensional axial and coronal fast-field echo T1-weighted imaging “black bone” sequences were scanned.
The obtained DICOM files were reconstructed in an axial and coronal plane for image analysis, with recon-struction layers of 40 and a thickness of 0.5 mm. Qua-litative and quantitative evaluations were performed by two board-certified radiologists. Imaging parameters included defect area in defined regions of interest and signal intensity value.
Harvesting and preparation of samples for histological and immunohistochemical examination
The animals were sacrificed at 2 and 4 weeks after the surgery. Rats were anesthetized with 10% chloral hydrate (4 ml/kg), perfused with 200 ml of normal saline, and then fixed with 200 ml of 4% paraformaldehyde in phosphate buffer. The right mandibles of each rat were excised and fixed with 4% PFA for 24 h. The mandibles were decalcified in 10% Ethylene Diamine Tetraacetie Acid (EDTA) at room temperature for 4 weeks. The decalcification solution was replaced twice a week. After complete decalcification, the bone tissues were dehydrated with gradient ethanol and immersed in paraffin for 2 h. The tissues were immersed in paraffin and sliced into 3- to 4-μm-thick sections. These paraffin sections were heated in a 62℃ oven for 20 min for HE or 2 h for immunohistochemical staining.
Histological staining, immunohistochemical staining, and immunofluorescence staining
Every fifth section of each group was stained with a hematoxylin and eosin kit (Beyotime, Shanghai, China). Samples were observed under a Ti microscope (Nikon, Tokyo, Japan) and measured using Image-Proplus 6.0 software (Media Cybernetics, Silver Spring, MD, USA). The percentage of newly formed bones was determined using the following formula: Newly formed bone (%)=(area of newly formed bone/area of the total defect)×100%.
Immunohistochemical staining was performed using the Biotin-Streptavidin HRP Detection System (ZSGB-BIO, Beijing, China) according to the kit instruction. Primary antibodies against hNUC (1:200), Runx2 (1:1,000), and OCN (1:500) were purchased from Abcam (Abcam, Cambridge, UK). Control sections were incubated with PBS instead of primary antibodies. Immunoreaction was detected with a diaminobenzidine (DAB) substrate kit (Maxim Biotechnology Co., Ltd., Fuzhou, China). Images were taken on a microscope (Nikon, Tokyo, Japan). The integrated optical density of Runx2 and OCN was calculated using the Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Before performing immunofluorescence staining, the sections were deparaffinized and blocked with 5% goat serum for 1 h. The sections were then incubated with primary antibodies against DMP1 (1:500), OCN (1:500), Runx2 (1:500), and Col II (1:500, all from Abcam, Cambridge, UK) at 4℃ overnight. Control sections were treated with PBS instead of primary antibodies. Samples were incubated with fluorescent-labeled secondary antibodies (1:500, Abcam, Cambridge, UK) for 2 h. Colo-calization of MIRB with DMP1, OCN, Runx2, and Col II was observed using a fluorescence microscope (Nikon, Tokyo, Japan).
Detection of the cytokines secreted by SHED
Cells were seeded at a density of 2×105 cells/well in a six-well plate. The next day, the culture medium was changed to a serum-free medium. The serum-free medium was collected after incubation with SHED for 0, 48, and 120 h. SHED-secreted cytokines were detected with the ELISA kit (Shenggong Biotechnology, Shanghai, China) according to the manufacturer’s instructions. A total of 100 μl of sample or cytokine standard was added to a pre-coated 96-well plate and incubated for 1.5 h at 37℃. The liquid was discarded, and the plate was dried. A 100 μl working solution of biotin-conjugated antibodies was added to each reaction well and incubated for 60 min at 37℃. The plates were rinsed four times with 1×washing buffer. A total of 100 μl of HRP-conjugated streptavidin was added to each well and incubate for 30 min to form an immune complex. The plates were rinsed four times. A total of 90 μl of substrate reagent (hiding from light) was added to each reaction well. The color developed at 37℃ for about 15 min. 50 μl of a stop solution was added to each reaction well to stop the enzyme–substrate reaction. The OD value at a wavelength of 450 nm was measured immediately using a microplate reader (within 5 min). The results were obtained by removing blanks from the standards.
Statistical analysis
Data were expressed as mean±standard deviation, and all data were analyzed using SPSS software (SPSS Inc., IL, USA). Comparisons between multiple groups were performed using ANOVA, and comparisons between two groups were performed using an independent-samples t-test. The statistical probability of p<0.05 was considered significant.
Go to :
Results
Isolation of stem cells from human exfoliated teeth and osteogenic induction analysis
SHED were isolated from the dental pulp of 6- to 8-year-old children as described in a previous report (Fig. 1A) (17). To evaluate the osteogenic capability of SHED, we cultured them in an osteogenic differentiation medium for 2 weeks. Alizarin Red S staining results showed that SHED were able to form visible calcified nodules after induction (Fig. 1B). Western blot analysis showed that the expression of Runx2 and Alp increased 10 and 4.5 times, respectively, after osteogenic induction treatment compared with non-treated control (Fig. 1C and 1D).
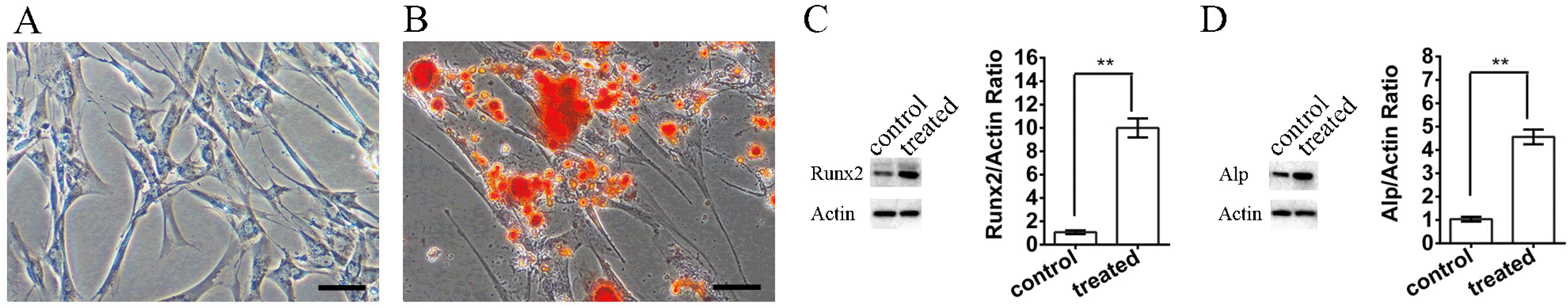 | Fig. 1SHED culture and osteoinductive treatment (A). The morphology of SHED isolated from the dental pulp tissue of children. (B) Alizarin Red S staining of SHED after osteoinductive treatment. (C, D) Changes in the expression of Runx2 and Alp before and after osteoinductive treatment. The scale bar indicates 100 μm.
|
SHED morphology, iron content, and live-cell ratio after treatment with various concentrations of MIRB
After incubating SHED with different MIRB concentrations for 24 h, the cells were stained with FITC-phalloidin to view their cytoskeleton and morphology, which showed green fluorescence. When the concentration of MIRB increased, there was no change in cell morphology, as showed by green fluorescence (Fig. 2A). The accumulation of MIRB in the cells was indicated by red fluorescence, which was emitted by MIRB particles them-selves. The red fluorescence of MIRB got brighter and stronger as the labeling concentration increased. This indicated that MIRB was accumulated more and more in the cells (Fig. 2A). Merged images of nuclei (blue), cytoskeleton (green), and MIRB (red) revealed that at varied labeling concentrations, the proportion of MIRB-labeled positive cells was approximately 100%. A similar result was obtained when the percentage of positive cells was calculated through Prussian blue staining (Fig. 2B).
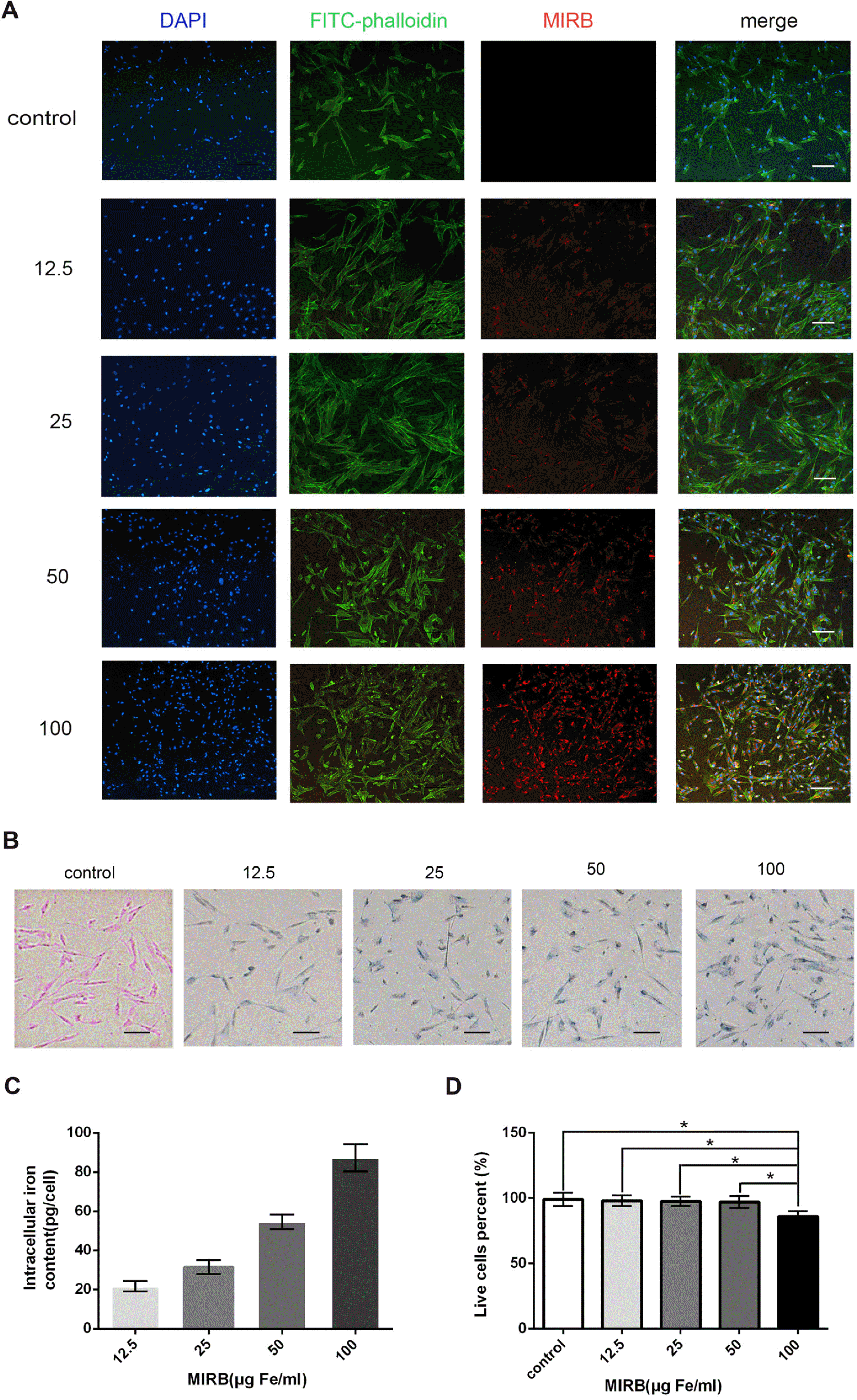 | Fig. 2Detection of morphology, iron content, and live cell ratio of SHED treated with various concentrations of MIRB (A). Morphology of SHED labeled with various concentrations of MIRB. SHED nuclei are stained with DAPI (blue); the cytoskeleton is stained with FITC-phalloidin (green); and MIRB particles are shown with Rhodamine B (red). (B) Prussian blue staining was used to detect Fe3+. (C) Iron content analysis of SHED. (D) Percentage of live cells was analyzed with Trypan blue staining. *p<0.05. The scale bar indicates 100 μm.
|
The iron assay kit was utilized to measure intracellular iron content fluctuation with increasing MIRB concen-tration. The findings indicated that the iron concentration increased as the labeling concentration increased, and the maximum iron concentration was observed at 100 μg Fe/ml (Fig. 2C).
Trypan blue staining was used to determine the effect of MIRB labeling on SHED growth by distinguishing between live and dead cells. The results showed that 100 μg Fe/ml MIRB reduced the percentage of live cells obviously compared with 0, 12.5, 25, and 50 μg Fe/ml MIRB (p<0.05). Other concentrations had minimal effect on the survival rate of SHED (Fig. 2D). Consequently, MIRB concentrations below 100 μg Fe/ml were considered safe for SHED labeling.
Establishing an appropriate MIRB concentration for SHED labeling
A cell growth curve was drawn to investigate the effect of MIRB concentration on SHED proliferation using the CCK-8 method. The results revealed that MIRB concentrations of 12.5, 25 and 50 μg Fe/ml had no effect on cell growth, whereas the MIRB concentration of 100 μg Fe/ml considerably inhibited cell growth (Fig. 3A). Therefore, 100 μg Fe/ml MIRB is unsuitable for in vivo labeling and tracking of cells and cannot be used to label cells in the following investigations.
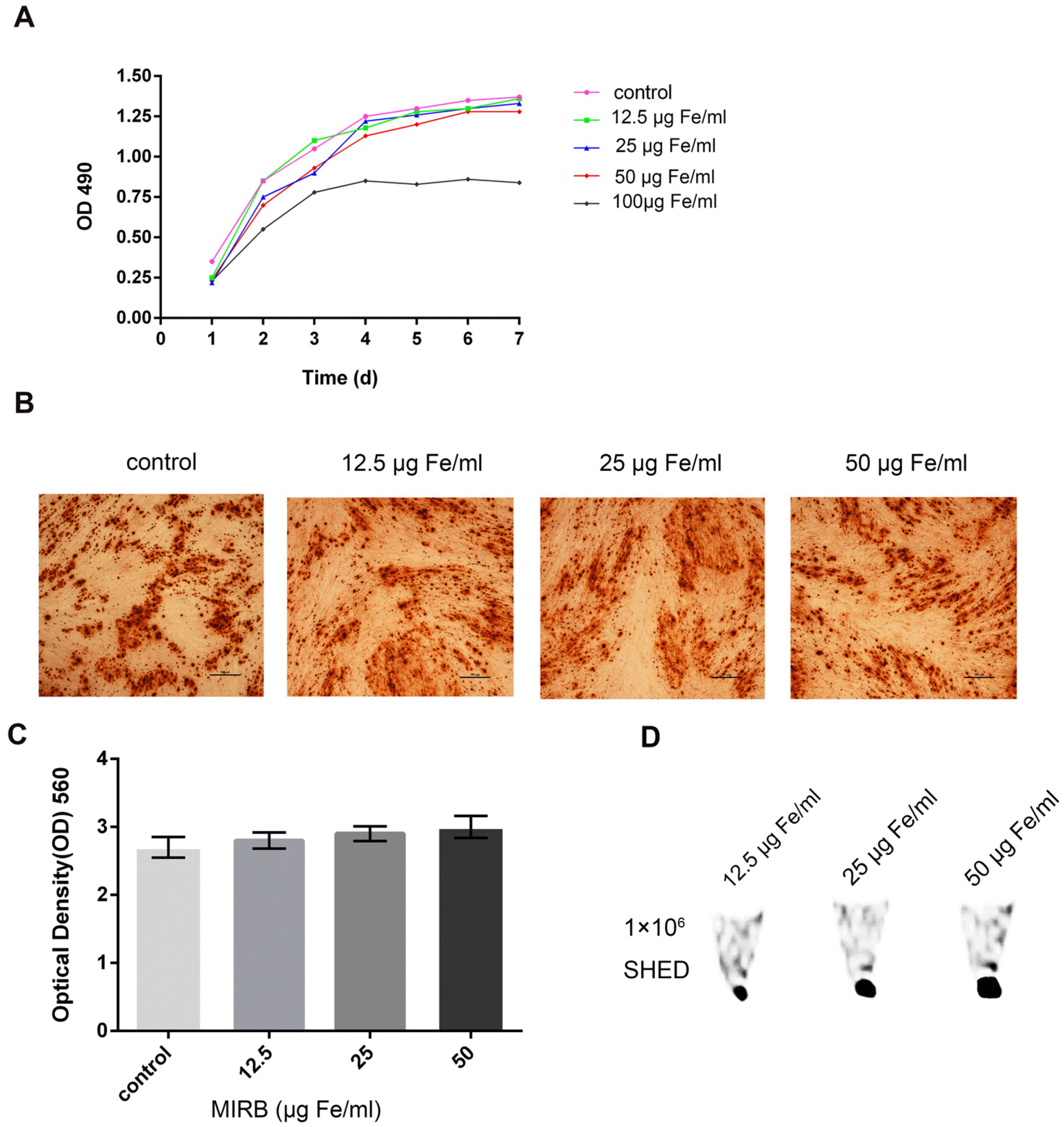 | Fig. 3Determination of the optimal MIRB concentration for use (A). Growth curve of SHED labeled with various concentrations of MIRB. (B, C) Alizarin Red S staining and quantitative analysis of SHED labeled with various concentrations of MIRB after osteoinductive treatment. The scale bar indicates 100 μm. (D) Sig-nal intensity images of 1×106 SHED labeled with various concentrations of MIRB in vitro by MRI.
|
We used Alizarin Red S staining and quantitative analysis to determine the effect of MIRB labeling on SHED osteogenic differentiation. The results revealed that the osteogenic differentiation potential of SHED labeled with various concentrations of MIRB (0, 12.5, 25, and 50 μg Fe/ml) is not significantly different (p>0.05, Fig. 3B and 3C). MIRB labeling concentrations of 12.5, 25, and 50 μg Fe/ml had no harmful effects on cell growth or differentiation, making them suitable for in vivo cell tracking.
The above experiments showed that MIRB is safe for cell proliferation and osteogenic differentiation in the concentration range of 12.5∼50 μg Fe/ml. To reduce the expense of the experiment, we should estimate the lowest MIRB concentration at which tracking through MRI is possible. Therefore, 1×106 cells were labeled with 12.5, 25, and 50 μg Fe/ml MIRB and resuspended in 15 μl of thrombin solution. Then, the mixture was combined with 15 μl of fibrinogen to form a gel for MRI examination. On spin-echo T2-weighted MR images, areas containing iron-labeled cells showed as low-signal-intensity regions, resulting in a negative contrast. The signal intensity increased as the MIRB concentration increased (Fig. 3D). Finally, 25 μg Fe/ml MIRB was chosen for in vivo labeling of cells.
Construction of periodontal bone defect and preparation of fibrin glue complex for SHED transplantation
Animals were randomly assigned to one of the following four groups: SHED (MIRB), SHED, Fibrin, or PBS. A 5 mm×4 mm×1-mm (length×width×depth) defect was created in each rat’s right mandible bone (Fig. 4A and 4B). Then, 15 μl of fibrinogen solution and 15 μl of thrombin solution containing 1×106 SHED were simultaneously injected into the defect region (Fig. 4C). The liquid turned into a gel in 1∼2 min. The muscle and skin layers were sutured separately.
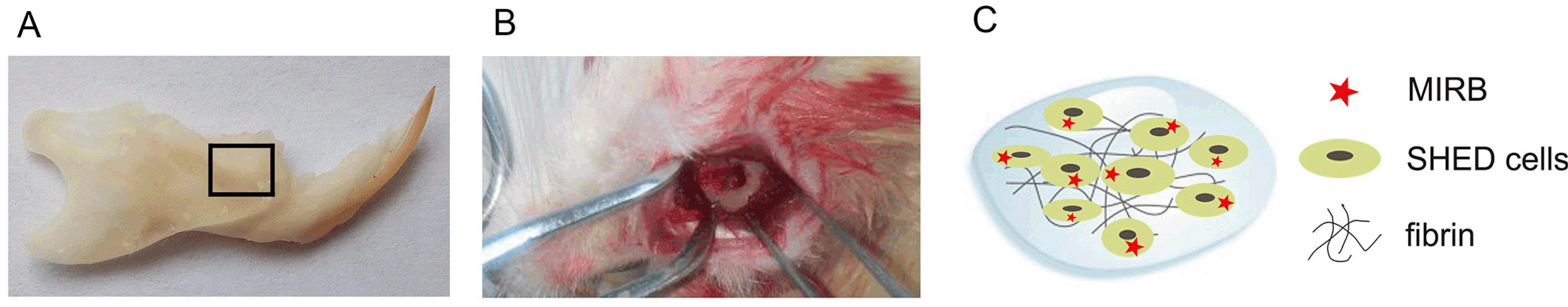
Determining the healing ratio of periodontal bone defects and tracking the survival of transplanted cells through MRI
MRI analysis was performed at 0, 3, 6, and 9 weeks after SHED were transplanted in rats. The bone healing percentage in the SHED (MIRB) and SHED groups was higher than that in the Fibrin and PBS groups at 3 and 6 weeks after the surgery (p<0.01, Fig. 5A and 5B). It is proposed that both MIRB-labeled and unlabeled SHED transplantation to periodontal bone defect is beneficial in repairing the defect. In the SHED (MIRB) group, the low-intensity artifact signal caused by Fe3+ can be observed at 6 and 9 weeks after surgery, as shown by the red arrows in Fig. 5A.
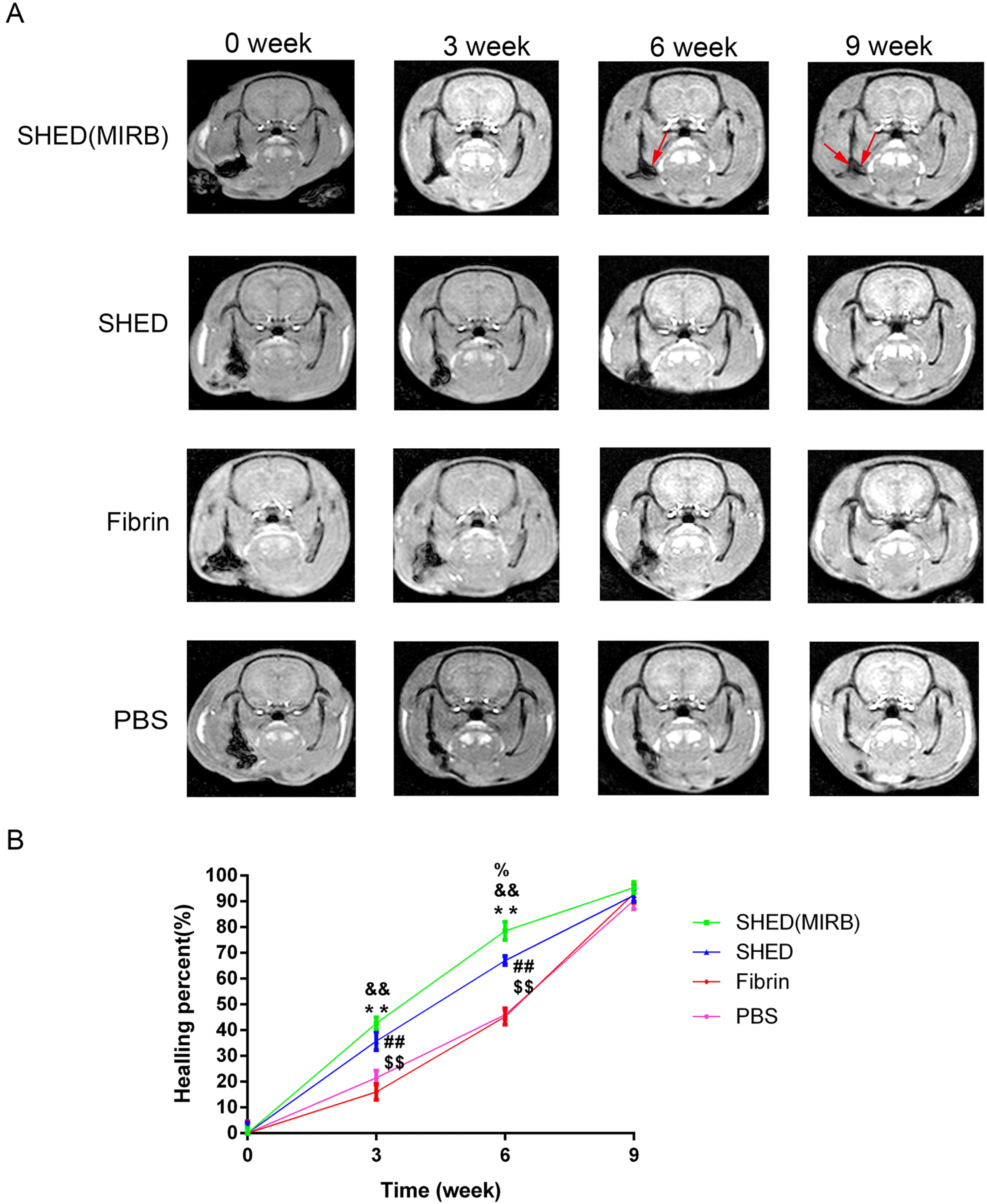 | Fig. 5MRI analysis of the healing ratio of periodontal bone defects and tracking the survival of transplanted cells (A). MRI images of rats at 0, 3, 6, and 9 weeks after surgery. Red arrows indicate the low-intensity artifact signal caused by MIRB. (B) The average healing percent of bone tissue at each time point was calculated according to the results of the MRI. Healing percent=(total defect area − present defect area)/total defect area. %,&,*: SHED (MIRB) group is compared with SHED, Fibrin, and PBS groups, respectively. #,$: SHED group is compared with Fibrin and PBS groups, respectively. %,&,*,#,$p<0.05. %%,&&,**,##,$$p<0.01.
|
Calculation of the healing percent of the defect with HE and immunohistochemistry
In addition to MRI, HE and immunohistochemistry were also used to analyze the percentage of bone tissue healing in vivo. HE staining and percentage of new bone in the defect showed no significant difference between the SHED (MIRB) group and SHED group 4 weeks after surgery (p>0.05; Fig. 6A∼B).The percentage of new bone in the SHED (MIRB) and SHED groups was significantly higher than that in the Fibrin and PBS groups (p<0.05, Fig. 6A∼B).
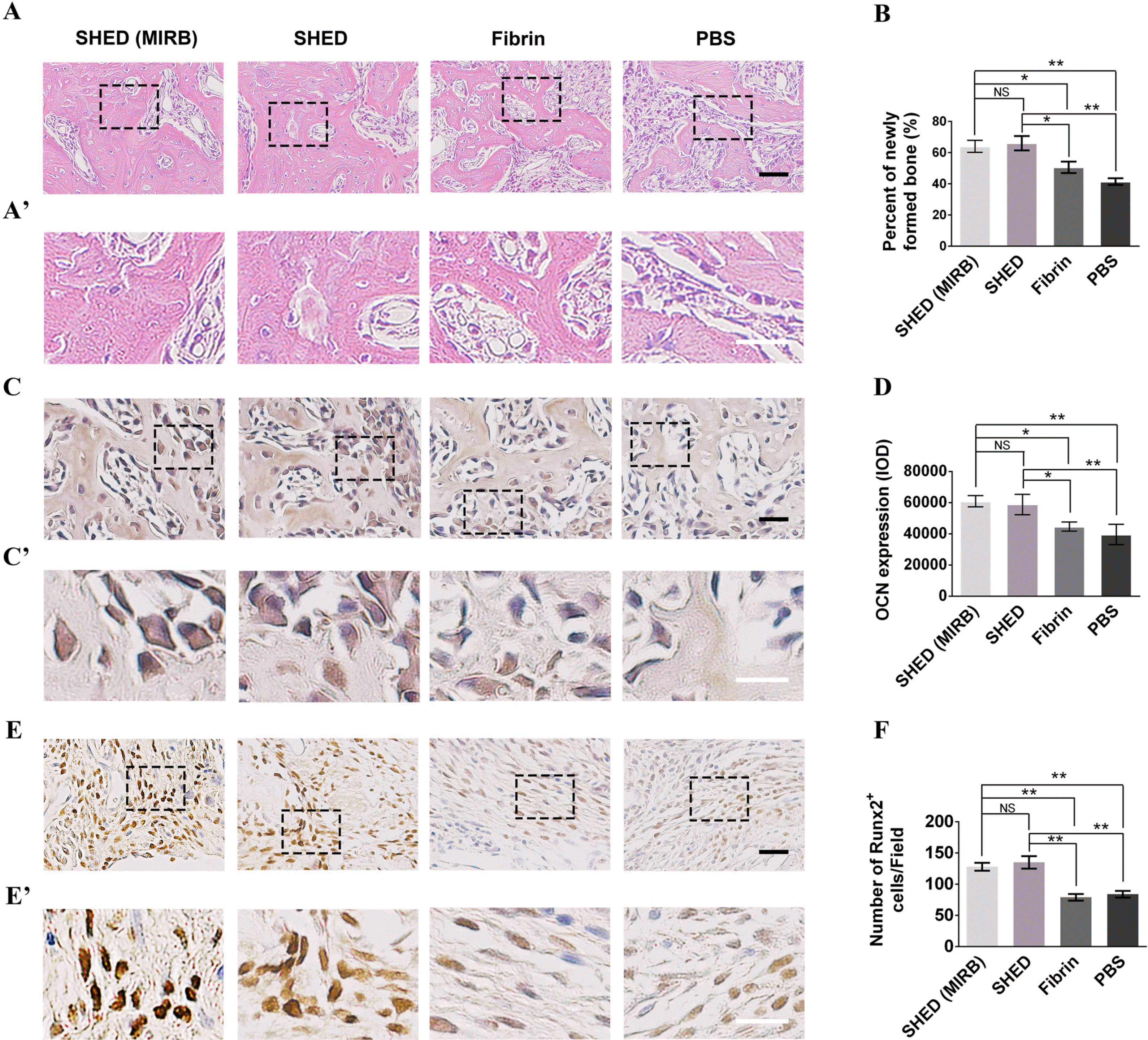 | Fig. 6The effects of SHED (MIRB-labeled and unlabeled) on bone regeneration (A). HE staining of the defected area 4 weeks after surgery. (A’) The higher magnified images in the boxed regions of (A). (B) Quantitative analysis of newly formed bone areas in four groups by HE staining. (C) Immunohistochemical staining of OCN 4 weeks after surgery. (C’) The higher magnified images in the boxed regions of (C). (D) Quantitative analysis of OCN expression in four groups. (E) Immunohistochemical staining of Runx2 2 weeks after surgery. (E’) The higher magnified images in the boxed regions of (E). (F) Comparison of the number of Runx2+ cells in four groups. The black scale bars represent 100 μm, and the white scale bars represent 50 μm. NS: not significant. *p<0.05, **p<0.01.
|
The optical density of OCN immunohistochemical staining was calculated 4 weeks after the surgery. The results showed that OCN expression in the SHED (MIRB) and SHED groups was significantly higher than that in the Fibrin and PBS groups (p<0.01) (Fig. 6C∼D).
Runx2 immunohistochemical staining was performed 2 weeks after the surgery, and the number of Runx2 positive cells was statistically analyzed. The number of Runx2 positive cells in the SHED (MIRB) and SHED groups was significantly higher than that in the Fibrin and PBS groups (p<0.01) (Fig. 6E∼F).These results indicated that transplantation of MIRB-labeled or MIRB-unlabeled SHED can promote bone tissue healing.
Survival and differentiation of SHED after transplantation in vivo
MIRB, a type of nanoparticle accumulated in the cytoplasm of labeled cells, has the potential to spread to neighboring host cells during cell division or apoptosis. To confirm that MIRB-labeled cells in vivo are indeed human SHED rather than host cells, we used the human cell-specific antibody hNUC for colocalization analysis with MIRB. The results revealed that all red MIRB fluorescence signals were found in hNUC-positive cells, whereas some hNUC-positive cells had no red MIRB fluorescence signal in their cytoplasm (Fig. 7A).This suggests that MIRB particles are lost in some SHED in vivo. MIRB particles are still present in some transplanted human SHED and can be used as tracer markers of SHED in vivo.
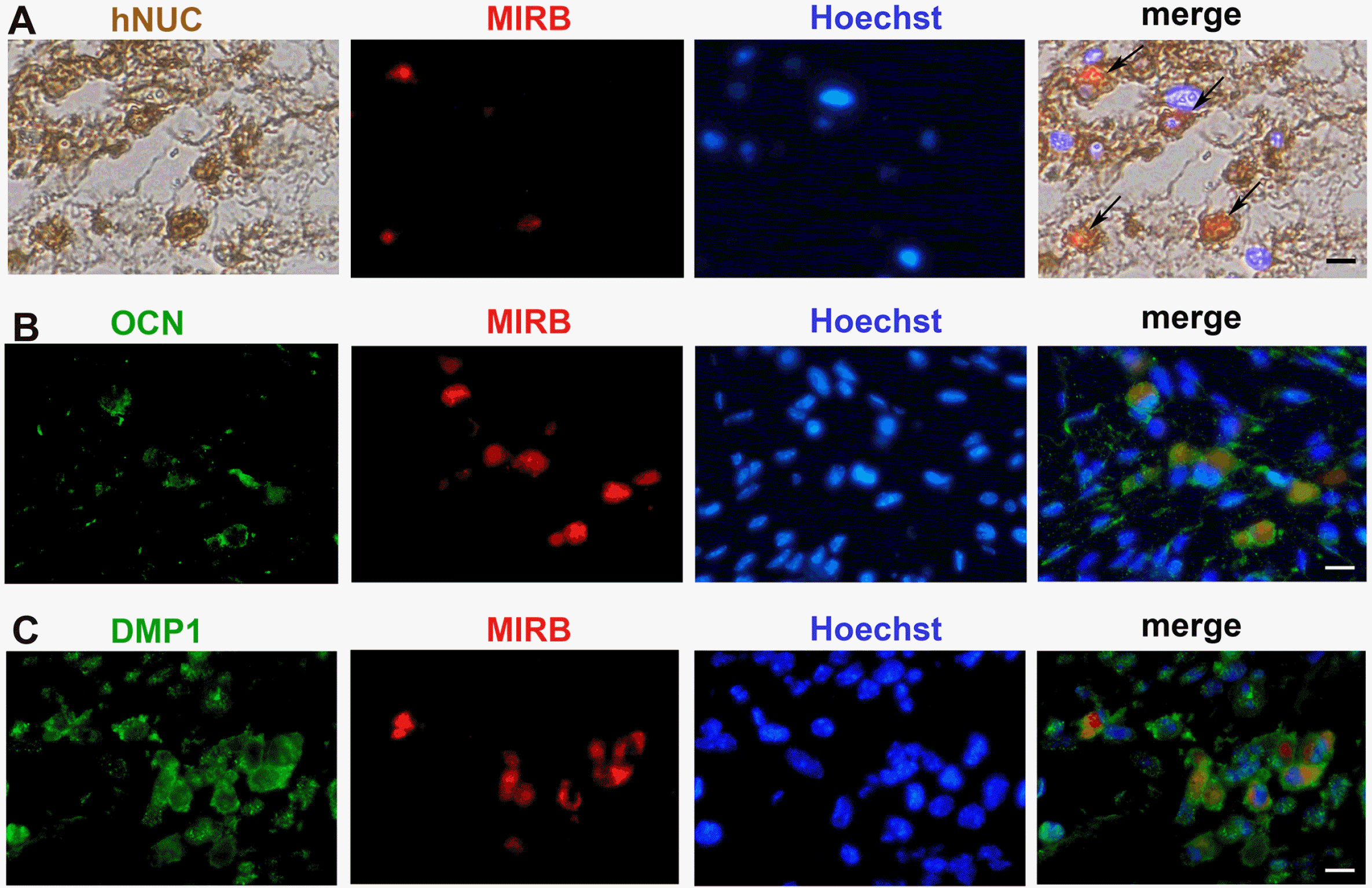 | Fig. 7Colocalization of MIRB and hNUC, OCN, and DMP1 in vivo (A). Immunohistochemical analysis of hNUC 4 weeks after transplantation. (B, C) Immunofluorescence colocalization of MIRB with OCN and DMP1, respectively, 4 weeks after the surgery. The position indicated by the arrow is the hNUC and MIRB double-positive cells. The bars represent 100 μm.
|
To investigate the cell types that SHED can differentiate into after transplantation, we performed colocalization of MIRB with OCN and DMP1 (Fig. 7B and 7C). Some SHED can differentiate into osteoblast-like cells and osteocyte-like cells that express OCN and DMP1, respec-tively. Consequently, we inferred that transplanted SHED are likely to promote bone regeneration through direct differentiation.
Protein factors secreted by SHED and the impact of transplanted SHED on their surrounding host cells
To determine if SHED could promote bone regene-ration through secretion of protein factors, we performed ELISA to detect the protein factors secreted by SHED that promote bone formation or inhibit inflammation (Fig. 8A). The results showed that SHED could secrete osteogenic factors such as OCN, ALP, and IGF-1. SHED could also secrete the protein factor IL-4, which suppresses inflammation; VEGF, which promotes angiogenesis; and bFGF, which promotes cell proliferation and survival. We deduced that the SHED might influence the surrounding microenvironment of the hosts by protein secretion after being transplanted into rats.
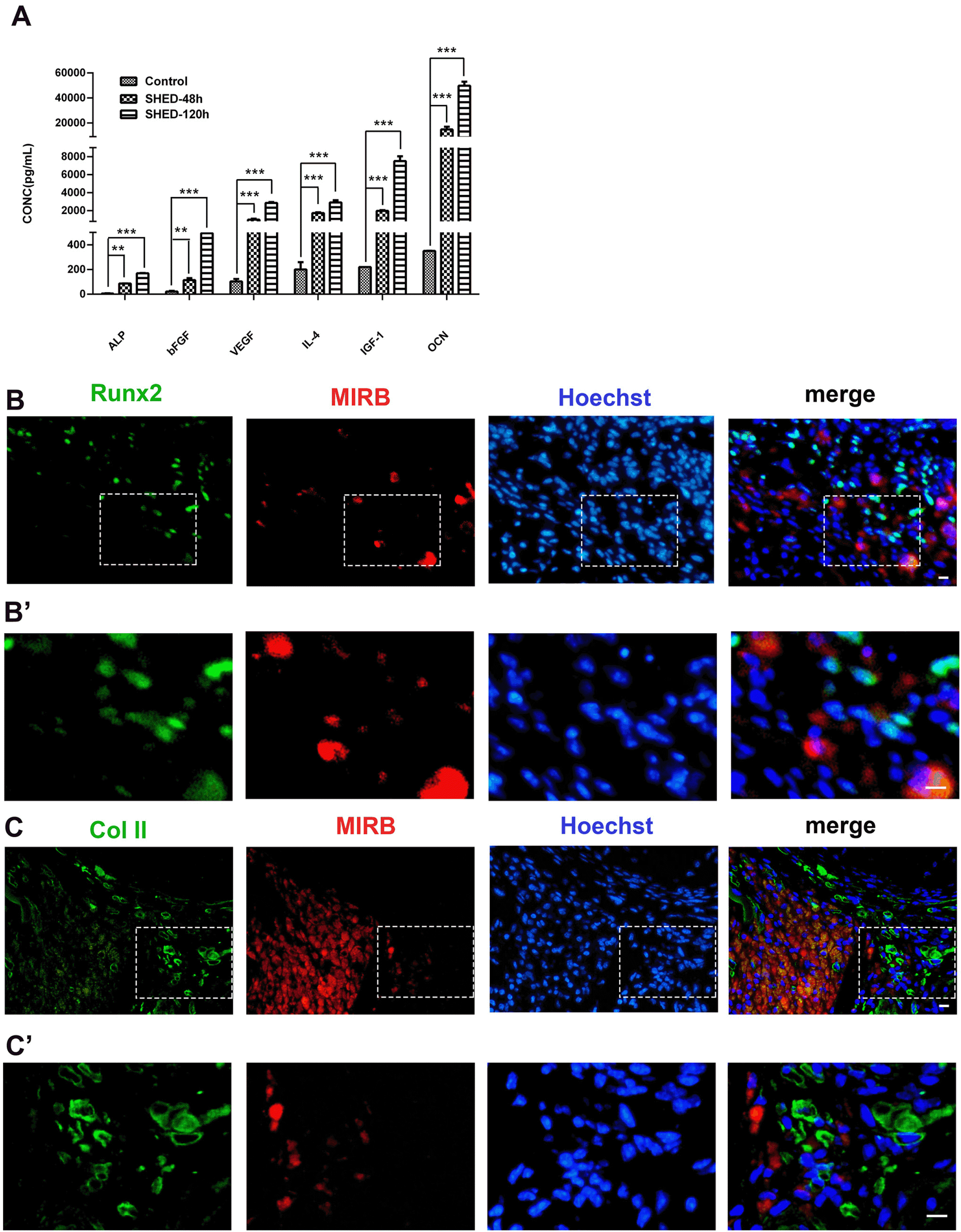 | Fig. 8Detection of protein factors secreted by SHED and the impact of transplanted SHED on their surrounding host cells (A). ELISA test of proteins factors secreted by SHED cultured in serum-free medium. **p<0.01, ***p<0.0001. (B, C) Immunofluorescence colocalization of MIRB with Runx2 and Col II, respectively, 2 weeks after the surgery. (B’, C’) The higher magnified images in the boxed regions of (B, C), respectively. The bars represent 100 μm.
|
In the colocalization analysis of MIRB with osteogenic proteins, a considerable number of host-derived Runx2-and Col II-positive cells congregated around the transplanted SHED was discovered (Fig. 8B∼C’). Runx2 is a pre-osteoblast marker and an important regulatory factor in the early stage of bone healing. Col II is a marker for hypertrophic chondrocytes and a key player in endochondral ossification during the bone repairing process. A large number of pre-osteoblasts and hypertrophic chondrocytes from the host congregated around the transplanted SHED, suggesting that the transplanted SHED can mobilize host cells through the paracrine to promote bone repair.
Go to :
Discussion
Alveolar bone destruction is a common hallmark of human periodontitis (18). In this study, we tried to transplant SHED into rats to repair the periodontal bone defect. The results showed that both MIRB-labeled and unlabeled SHED could promote bone tissue regeneration. MRI analysis revealed that MIRB-labeled SHED could be tracked for 9 weeks in vivo. Immunofluorescence staining showed that some SHED could differentiate into osteoblast-like and osteocyte-like cells. However, most SHED did not differentiate or migrate in vivo. In addition, a large number of endogenous rat pre-osteoblasts and chondrocytes gathered in the peripheral areas of the transplanted cells. SHED could release osteogenesis-related substances such as OCN, ALP, IGF-1, IL-4, VEGF, and bFGF to alter the microenvironment of the transplanted area. These findings implied that SHED promotes bone repair through direct cell differentiation and paracrine mobilization of endogenous rat cells.
Stem cell tracking is an important method for revealing the mechanism of stem cell therapy by indicating the distribution and differentiation of transplanted cells (19). Many tracking methods, including MRI, fluorescence ima-ging, and ultrasound imaging have been developed (20). In this study, we used MRI to track cells in vivo by labeling SHED with a SPIO negative contrast agent. A new SPIO agent Molday ION Rhodamine-B (MIRB) was used to track the transplanted SHED. MIRB is labeled with Rhodamine B, a fluorescent dye, and can be visualized by both MRI and fluorescence imaging (15, 16, 21, 22). However, as a direct cell labeling approach that relies on the accumulation of nanoparticles in the cytoplasm, MIRB has some technical flaws. First, high concentrations of intracellular free iron are toxic to the cells. Our results indicated that MIRB concentrations more than 100 μg Fe/ml had a negative impact on cell viability and proliferation, which is in line with previous research (16). A MIRB concentration of 25 μg Fe/ml was employed to fulfill not only the signal intensity required for MRI detection but also to have no detrimental effect on cell proliferation and differentiation. Second, the labeled nanoparticles will be lost during cell division, resulting in weakening of the MRI detection signal (23). At 9 weeks after the surgery, MRI images revealed that the rat’s periodontal bone defect had healed completely, but there were still low-density artifacts at the transplant site produced by MIRB. Previous research found that the MIRB-labeled signal can last for 60 d and 42 weeks in different studies, but the MRI signal gradually fades over time (16, 24). This demonstrates that, although MIRB is lost to varying degrees after transplantation, it can still be used as a reference for the survival of transplanted cells in vivo. Third, apoptosis and macrophage phagocytosis create leakage of MIRB particles from labeled cells, which are readily absorbed by nearby host cells, resulting in false-positive results (20). To determine whether the low-density artifact was generated by MIRB-labeled SHED or MIRB overflow-labeled rats’ cells, we performed immunohistochemical staining with the human cell-specific antigen hNUC. The findings revealed that all cells with red MIRB fluorescence were positive for hNUC histochemistry, not all hNUC-positive cells had red MIRB fluorescence. This demonstrated that MIRB was lost with cell proliferation after cell transplantation in vivo. However, a significant number of transplanted cells were still labeled with MIRB, and MIRB tracking can still reflect the survival of transplanted cells in vivo.
In recent years, stem cell transplantation has emerged as one of the most promising approaches for bone regeneration therapy (25). However, the stem cell transplantation mechanism for bone regeneration therapy remains questionable. There are two main points of view. The first is that stem cell transplantation repairs bone defects through direct differentiation, whereas the second is that stem cells promote bone healing through paracrine pathways (26). Previous findings have proven that the transplanted cells could differentiate into osteocytes and deposit bone matrix proteins such as OPN and OCN to induce new bone formation in the grafted defect area (27). Our study revealed that transplanted SHED can differentiate into OCN-expressing osteoblast-like cells and DMP1-expressing osteocyte-like cells, implying that the transplanted cells can participate in bone healing through direct differentiation. However, a large number of Runx2- and Col II-positive cells were discovered around the transplanted cells. Runx2 is a pre-osteoblast marker and a critical regulator in the early stages of bone repair (28). Col II is a hypertrophic chondrocyte marker and a key player in the endochondral ossification process during bone repair (29). The transplanted SHED attracted large numbers of host pre-osteoblasts and hypertrophic chondrocytes, suggesting that the transplanted SHED can mobilize the host cells through paracrine to promote bone healing. These findings suggested that SHED promoted bone repair via direct differentiation and paracrine processes.
In the future, we should focus on deciphering the signaling pathways governing SHED destiny selection after implantation, as well as the molecular dialog between SHED and host cells in vivo. Determining the mechanisms by which SHED repair the bone defect, whether through direct differentiation or paracrine pathway, is indeed essential for future progress in tissue engineering strategies.
Go to :
 XML Download
XML Download