

 PDF
PDF Citation
Citation Print
Print


Introduction
Historically, science has always been a battle against uncertainty. Humanity has accumulated new knowledge by approaching the unknown in a way that is considered the best at the technological level of the time. Therefore, knowing what is presently best and recognizing the reliability and limitations of such methods are fundamental requirements for developing new science. Cell therapy (CT) products belong to the cutting-edge area of medical science (1-3). Although research in the field of CT has been conducted for a long time, developing it as a therapeutic agent applied to actual patients is a completely different task (4, 5). This is supported by the fact that there are few CT products approved worldwide so far, and the development of CT products means that we are faced with a situation we are not yet familiar with (6, 7). Therefore, to properly develop CT products, understanding the current consensus in the development of new drugs and how this is applied to the uncertainty in the field is necessary. Furthermore, clearly recognizing the limita-tions of such a methodology and having a logic to respond to it should be possible.
Many stakeholders lack an understanding of the mechanisms and pathways of drug development, and the plan for generating evidence for various decisions inevitably encountered during the development process is not robust in many pipelines (8, 9). This is the first difficulty that many sponsors are facing in developing CT products. CT does not require a logical justification that is completely different from that of conventional drugs however, it needs more fundamental evidence for safety and efficacy. Furthermore, the level of evidence is not determined in such a way that “there must be specific experimental results” (10). In this regard, based on the latest consensus, the question “is the best evidence secured?” may be appro-priate. Simply checking the existence of data is not sufficient to answer this question. Instead, it requires a high-level task based on a comprehensive understanding of the whole development rationale.
In this context, finding the most accurate evaluation methodology for investigational products (IPs) is another general problem. This is where CT products clearly differ from classical drug products. Small-molecule drugs or therapeutic antibodies have a long history of development and approval, and relatively sufficient experience has been accumulated. This includes what information should be secured through what experiment and how to fuse such information into knowledge about the IP. However, in the CT field, the characteristics and functions of cells to be developed are diverse thus, the type of experiment and evaluation items for each product must be customized. Therefore, having the ability to judge which evaluation result can be said to be the best at the current scientific level and how it can be extrapolated to the next development stage is necessary. Furthermore, a technique for preventing the developmental timeline from being extended by accurately arranging the start time in consideration of the execution period of individual tests is needed.
In this article, the current status of CT product development was analyzed, and major unmet needs were identified. Furthermore, to solve such a problem, the types of evidence for each stage that should be prepared for developing CT products were discussed compared with the consensus of classical drug development. Finally, measures were proposed to strengthen the competitiveness of CT product development in the future.
Trend in CT Product Development
Increasing complexity as a drug product
Developing CT products inevitably requires clinical trials. Despite the tremendous scientific advances in the field of CT, there have not been many approvals as a therapeutic agent until recently because of the difficulties at this stage. Naturally, products expected to have low risks when applied to humans in terms of cell characteristics, route of administration, and distribution in the body have reached marketing approval earlier (6). Another consideration that must be considered is the therapeutic benefit. Even those belonging to the relatively safe class of CT are considered at a higher risk than small molecules. Therefore, development has been progressing from indications that CT can show dramatic improvement in treatment outcomes compared with existing treatments. Table 1 presents the characteristics of various CTs classified according to the risk-to-benefit ratio (11-13). In this context, most CT pro-ducts first applied to clinical trials (with some exceptions) had limited distribution in the body, such as the skin and cartilage, and showed local efficacy (6, 14-16). After these products, autologous cell-derived somatic cell products for systemic administration reached marketing approval.
Implications of recent CAR-T approval
The approval of Kymriah (tisagenlecleucel, Novartis) and Yescarta (axicabtagene ciloleucel, Kite Pharma/Gilead), autologous chimeric antigen receptor-T (CAR-T), in the United States and Europe indicated that an international consensus on the regulation of autologous CT was established (17, 18). Immune cells inevitably exhibit systemic functions and belong to a relatively high-risk somatic cell therefore, regulatory standards for these products can serve as a framework for autologous somatic cells with a lower risk. Moreover, this was significant in that it presented a standard for risk assessment of systemic CT products to which genetic manipulation technology was applied.
Clinical development and licensing of autologous CAR-T has since facilitated the development of similar products (19-21). Refractory cancer still presents a good opportunity for these products because the therapeutic benefit of most new treatments is considered to outweigh the risk. Existing CAR-T has mainly focused on hematological malignancies due to cellular characteristics and some biological limita-tions. Of course, compared with existing anticancer therapy, the performance was dramatic however, many sponsors believe that they can develop a product that can show efficacy even for patients refractory to existing CT therapy by improving the performance of autologous CAR-T (21). Such improvements are leading to attempts to apply autologous CAR-T to solid tumors (22, 23). Particularly, cancer types with poor treatment responses to existing chemotherapy are the main targets. Furthermore, it naturally aroused interest in whether CT products with immune function can be used allogeneically rather than autologously (24, 25). In terms of commercialization, allogeneic products have superior advantages in production and management compared with autologous ones. Although allogeneic administration has a higher risk than self-administration, a certain level of consensus on the risks of graft-versus-host disease (GVHD) and immunogenicity was formed through various biological knowledge and clinical expe-riences. Recently, various clinical trials of allogeneic CAR-T products have been conducted, and the development of CT products using natural killer (NK) cells, which do not cause GVHD, is also active. In the next decade, it is expected that various CT products using autologous and allogeneic somatic cells will make up most clinical development and product approvals.
What about stem cells?
CT using stem cells has a relatively high-risk compared with other categories due to the following problems related to the uncertainty of cell differentiation after administration to the human body (11-13):
Unwanted immune reaction
Genetic instability and tumor formation
Dedifferentiation/loss of function
Unintended alteration of cell homeostasis
Unwanted ectotrophic engraftment/biodistribution
Up to now, stem cell products have been mainly developed as a topical formulation with relatively less concern than systemic administration. The condition of topical administration has a limitation that it should be able to easily reach the diseased site even using a noninvasive method. Therefore, in the beginning, its indications were limited to the skin or large joints however, with the accumulation of knowledge for more than a decade, it is now challenging disease areas with a higher risk. Recently, the major interest in stem cells is the field of irreversible cell damage. Eye-related clinical trials as unmet medical needs are the most frequent, followed by more life-threatening diseases, such as stroke and myocardial diseases (26). Cells can be delivered directly to these tissues using various methods, such as direct injection and vascular intervention, which do not damage the tissue. Furthermore, since the therapeutic benefit is greater than the combined risk of CT itself and the administration method is expected, such an attempt can be justified. For example, the clinical development of dry age-related macular degeneration treatment using embryonic stem cell-derived retinal pigmented epithelial cells is also in progress (26). More stem cell products are entering the field that have a higher risk in terms of the possibility of exposure to systemic circulation, such as brain and heart diseases (27-29). To support the development of stem cell products, a common standard for the level of requirements for the clinical development of such products is being sought. “Stem Cell-Based Clinical Trials: Practical Advice for Physicians and Ethics/Institutional Review Board” recently proposed by the International Society for Stem Cell Research is a good example of risk/benefit evaluation items related to the first-in-human (FiH) admi-nistration of stem cell CT products (30).
Summary for general trend
The era in which CT is considered a treatment option in the actual clinical field has arrived. Accordingly, it is expected that various studies on CT and its clinical development will become more active. Given the current trend, it is only a matter of time before the actual clinical use of stem cells increases. However, CT remains a relatively new field, and many pipelines are in the preclinical development stage. Many sponsors are now accumulating development experience. The expertise required primarily in the current situation is related to the generation of evidence for initiating clinical development and the methodology for designing an early-phase clinical trial based on it. For this purpose, understanding the requirements of the evidence that IP should have for clinical development is necessary, and explaining the rationale required for CT and the reason through considering the inherent difference between classical drugs and CT should be possible. In the following section, essential contents related to this will be concisely discussed.
General Logic to Justify Clinical Development
Similarity in rationale for initiating clinical development
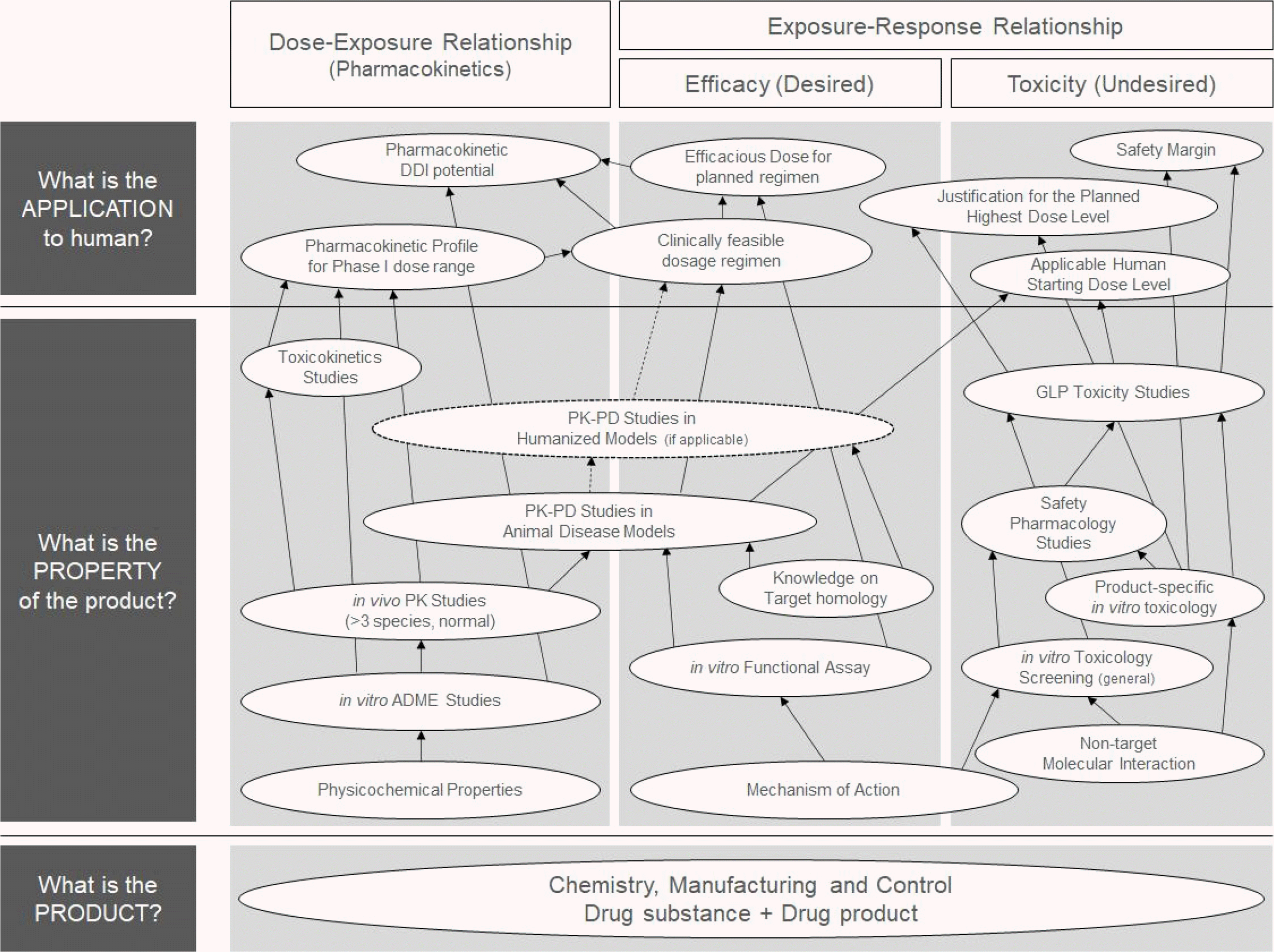
The level of knowledge on IP required for initiating clinical development is not much different from that of classical drugs for CT products (31, 32). Therefore, understanding how to combine various pieces of preclinical information to create a rationale for the clinical development of classical drugs is important for CT sponsors. This can be summed up in one sentence: “Is the risk to be taken by the subject acceptable given the expected benefits?” Since these benefits and risks are evaluated based on the basic pharmacological evidence of dose-exposure–response, the three large domains, pharmacokinetics (PK), efficacy, and toxicity, are key information elements. The overall process of generating and using generally applied preclinical evidence is expressed as a diagram (Fig. 1). However, the arrows in the diagram are in a logical order and do not necessarily indicate that tests to obtain such information must be performed sequentially.
Identity of the drug product: chemistry, manufacturing and control
Since the subject of this article is the preclinical basis of CT, the meaning of chemistry and manufacturing and control (CMC) should be discussed before discussing the properties of pharmaceuticals. This is a basic element allowing information derived from different studies at different times to be combined and used. The CMC document describes the definition of the product, the production process, and the procedure to check whether the output is the defined product (32). If there is a change in the production process, the output can be changed therefore, the production process can also be a part of the identity of the IP. It is defined for both active ingredient (i.e., drug substance (DS) and final formulation (i.e., drug product (DP) levels. If several studies are conducted using the IP manufactured usingdifferent methods in a state where CMC has not been established, grouping and interpreting the results derived from those studies may be difficult. For example, linking and interpreting the PK data obtained from a study administered with DS solution even at the same dose and in the same species and the pharmacodynamic (PD) data observed after DP administration is not advisable. This is because, even if the DS is the same, the drug exposure will change if the administered formulation is different. A clear definition of DP for human administration and preclinical data obtained using it contribute greatly to the validity of initiating clinical development.
Regardless of the classification of drugs, establishing the CMC for DS and DP as early as possible and formulating a development plan while considering the specifics are recommended (32, 33). It should be emphasized that establishing a CMC is distinctly different from obtaining a CMC document for submission to a regulatory body. CMC documents submitted to the regulatory body must be of a quality acceptable to the regulatory body. This means that the document should be structured in the form of an SOP that has the proper format and content so that anyone can perform the procedure. The production of internally established production and quality control procedures into these documents is also time-consuming. Thus, if there is a planned clinical development initiation timeline, the preparation of the CMC document should begin at least several months in advance.
Value of nonclinical dose-exposure information
PK factors define the relationship between dose and in vivo exposure. Considering that the human body’s response to a drug is formed in proportion to the level of exposure, not the dose itself, it must be confirmed before efficacy or toxicity. Because the PK is not an emphasized information for CT products, to summarize only the core of the PK evidence required for clinical development, it can be said that “the best possible PK evidence for possible exposure levels in humans is required” (34). The reason that this scientific demand for classical drugs is possible is that human PK can be reasonably predicted using various pieces of in vitro information and in vivo exposure results together. The value of various in vitro and in vivo tests for predicting human PK in classical pharmaceuticals is justified due to the experience and knowledge accumulated over a long time. Conversely, if the preclinical observations do not have the value to be extrapolated to humans, various in vivo studies cannot be justified considering animal experimentation ethics.
Evidences for efficacy
Evidence regarding efficacy is obtained from various in vitro and in vivo studies (35, 36). Considering that target binding is essential information in the stage of screening various candidate substances, if it is a substance under development, some PD properties have been confirmed. However, this information does not constitute a proof-of-mechanism about whether an intended change is caused in the target through such a combination. Therefore, the basis for causing an intended physiological change even at the cellular or tissue level is a basic requirement for both drug development itself and licensing-in or licensing-out. Going one step further, whether the intended change in the target ultimately represents the desired clinical result (Proof-of-Concept) is a key basis for predicting development success. To observe clinical results, the following two conditions must be met. First, there should be in vivo study results. Second, the animal model of such a study should be etiologically and clinically comparable to the actual human disease state. Therefore, in developing classical pharmaceuticals, the existence of relevant animal models is considered an essential factor. Animal PK–PD studies, which can be said to be a key step in the preclinical stage, are conducted for these models (37, 38). The dose range to be used in the study can be determined by combining the drug concentration that showed a significant action at the in vitro level and the in vivo PK results in the animal model species. Unfortunately, disease states induced in animals are often fundamentally different from those in humans, which is often cited as a limitation. This is a major problem that reduces the accuracy of human PK–PD prediction using the results of the study. Recently, the development and use of humanized animal models or in vitro-based human-simulating systems (e.g., microphysiological systems) is increasing (39, 40).
Evidences for toxicity
Lastly, toxicity-related evidence can be the most essential information for clinical entry in some respects. Since the regulatory framework related to the investigational new drug (IND) originates from an event related to drug safety, the current review for clinical development initiation also primarily focuses on securing safety. In this context, for the key evidence of safety, reliability is strongly emphasized that only results performed in facilities that meet specific requirements—Good Laboratory Practice (GLP)—are accepted (41). This fact sometimes leads to the misconception of some sponsors that only GLP-toxicology studies should be conducted in preparation for the IND. Also, the fact that safety is a key rationale is no exception to CT however, this does not mean that a GLP-toxicology studyis always required.
Although a GLP-toxicology studylooks at some macroscopic health indicators, it prioritizes histopathological findings (42). Since these changes appear differently depending on the animal species, dose levels, and observation time for each endpoint, in vivo non-GLP exploratory toxicology studies must be sufficiently performed before obtaining accurate GLP-toxicology results. Furthermore, safety pharmacology, such as proarrhythmic potential and cytotoxicity (especially for vital organs), is performed. Before that, in vitro toxicity screening constructs the basis for all other toxicity studies. This work was also designed to secure basic requirements for clinical development by eliminating candidates that are not “drugable”. From this, evidence of toxicity is sequentially accumulated. The reason that this entire scheme is possible is that nonclinical study results of various levels have a value as basic information that can predict toxicity in humans. However, such a predictability may not hold for CT. Table 2 shows the regulatory and scientific concerns regarding the IND as a summary of contents discussed so far.
Preclinical–Clinical Translation of CT Products
Inborn nature of cell therapy products distinguished from small molecules
The questions to confirm the feasibility of clinical development and the appropriate level of answers are similar, regardless of the type of drug. However, for CT, the supporting evidence for the answers is different because it is fundamentally different from conventional pharmaceuticals in its properties (Table 3). By considering the contents of Tables 2 and 3 together, roughly grasping how to prepare the basis for initiating the clinical development of CT products is possible.
Functional aspects of product identification
First, there is a discrepancy in the methodology for addressing the question “What product is administered to the human body?”. Since conventional pharmaceuticals define an active ingredient based on molecular structure, it can be proven that the produced substance is an active ingredient using various qualitative/quantitative analysis techniques. In contrast, many biologics require this definition in terms of structure and function. Before CT, this dimension of approach has been made using the concept of “totality of evidence” in developing biosimilars. CT is no exception, and since cells are much more complex than therapeutic antibodies, only structurally defining them is limited. Sponsors should clearly specify the nature and origin of the cells used for CT, as well as the manipulation methods and materials applied to the cell. Simultaneously, various types of tests to evaluate cell function and criteria to prove whether the minimum performance as a CT product is secured should be presented. These evaluations include not only items on cell efficacy but also items on whether substances or ingredients with the potential to cause toxicity (e.g., viral vectors and mycoplasma) are properly removed. One advantage of CT products over chemical pharmaceuticals is that they do not require formulation as a special drug delivery device. If only the characteristics of DS are well defined, the final product can be defined only by considering conditions related to logistics and storage. Therefore, to minimize additional work during the development, defining an appropriate evaluation plan from raw materials, intermediates, and finished products as early as possible is recommended.
Product characteristics and possible concerns
Although defining the IP of CT is more difficult, the benefits obtained through such a definition process are superior to those of conventional drugs. For conventional drugs, the definition of IP does not mean more than what is administered to the human body has been confirmed, and various characteristics related to the interaction with the physiological system should be evaluated using various in vitro/in vivo tests. Recently, various computer-based techniques for finding molecules with optimal target and nontarget interactions have been widely used however, fully identifying a series of physiological cascades occurring following molecular interactions is impossible. In contrast, CT is developed using a relatively specific rationale based on extensively researched biological knowledge. Therefore, if the characteristics of IP are clearly defined, it can serve to some extent as a predictive basis on the cell distribution and biological action in the human body. Therefore, the CMC of CT is an essential document for defining the expected risks and benefits of an FiH trial. This will determine the key questions regulatory agencies may have about safety and will change the rationale for the clinical development that should be prepared accor-dingly. Thus, the evidence to support the feasibility of clinical development is product-specific. Since this type of risk/benefit analysis has been applied similarly to other biologics, various clues can be obtained for developing CT products by referring to such cases. In this respect, establishing CMC in CT is a critical part of preclinical development and is a high-priority task. In many approved CT examples, the CMC-related section and discussion comprise a major part of regulatory documents even at the marketing approval stage (43-45). As the CTs are sequentially developed according to their potential risks, examples for the most well-described CT characteristics, safety concerns and their solutions can be found from the recent approval cases - the CAR-T products (17, 18).
Difficulties in the nonclinical evaluation of ‘partial’ intrinsic human components
The fact that securing evidence for the clinical development in a product-specific manner is necessary causes many difficulties in the preclinical development (46). This means that we remain more familiar with the logic of developing conventional drugs, in which the framework of preclinical evaluation is relatively stereotyped. This is possible because conventional drugs are similar that they are “xenobiotics” for both humans and animals, and thus, information (particularly for safety) obtained from animals can be reasonably extrapolated to humans. Many in vivo studies for drug development are justified for this reason, despite ethical concerns, and are considered a key step for nonclinical–clinical translation. The feasibility of the selection of the starting dose in humans and escalation design is also supported by various applications of the evidence. Therefore, the accurate and effective conduct of in vivo studies is crucial, and for this purpose, preclinical candidates should be selected, and appropriate dose range and endpoint observation period should be defined through extensive in vitro and exploratory studies. However, at the current scientific level, CT primarily refers to therapeutics using human cells. Naturally, the administration of CT to other species will produce different results from its administration to humans. This includes various immune responses, discrepancies in cell distribution, proliferation and differentiation, and the differences in target interaction. It cannot be assumed that the insufficient efficacy and/or overwhelming toxicity observed in animals will also apply to humans. To overcome this limitation, there are cases where an in vivo study is conducted using an animal cell manufactured to have the same mechanism as the IP (mock cell). Examples of efficacy studies using this scheme can be found in the nonclinical pharmacology section from the regulatory review document of approved CTs (45) and related articles. However, considering this as direct evidence to support the clinical development is difficult since it does not use actual IP. In the same context, the results of the GLP-toxicology study cannot be a basis for key decision-making. Overall, the value of in vivo studies in CT product development is significantly lower than that in the development of classical drugs, and product-specific evidence should be obtained through a combination of various information rather than direct evidence in this narrow range.
Emphasis on communication and consensus building
Presenting the problem of what information and how to combine it to create a valid basis for clinical development as a simple principle is difficult. Therefore, continuous discussion based on CMC between the sponsor and regulatory agency is necessary (47). Looking at the development cases so far, it is necessary to prove that the general regulatory requirements for product quality are secured with the CMC document. Furthermore, based on existing biological knowledge, extensive in vitro test results are needed to determine whether the cells themselves and the manipulations applied to them produce the intended results. This includes, but not limited to, the direct result of manipulations applied to the cell, the proliferation and differentiation of the cell, and the intended or unintended function of the cell. The general characteristics (i.e., normal physiological number, distribution, and function) in the human body of the origin cells that have not been manipulated, the clinical use experience of CT using the same cell or the result from sporadic compassionate use, among others, can constitute a rationale. For example, the human starting dose of CT products is often mainly supported by such knowledge, and if the characteristics of products are similar, the starting dose tends to be similar. The starting dose for CAR-T is between 106 and 107 cells/infusion, and for NK products, the starting dose was between 108 and 1010 cells/infusion. Furthermore, although having an ancillary value, the rationale can be further strengthened by conducting an in vivo test simultaneously measuring exposure/efficacy/toxicity endpoints in an animal model using settings as similar as possible to the human body. A multidisciplinary approach is essential to establish the basis for clinical development of CT products. However, even if all information is combined, the level of evidence is lower than that in conventional drugs. Since accurately predicting the cellular kinetics, unknown biological activity (e.g., carcinogenicity and cytotoxicity), potential for differentiation into other cells, and ability to proliferate beyond expectations in the human body is difficult, a more conservative approach is desirable if there is a lack of experience in the clinical use of similar CT products. The feasibility of FiH can be improved by applying a larger safety factor (e.g., 30∼50), an acceptable dose-escalation plan (an increase that does not exceed half-log [approximately thrice higher than the previous cohort] has been recommended), and a closer safety monitoring plan. The maximum planned dose in FiH should be higher than the predicted effective dose; however, since finding the maximum tolerable dose is not a primary concern for most CT products, it should not be exceedingly high. For this reason, the dose levels were ≤3 in many CT FiH trials. Moreover, even in the case of a FiH trial, considering that the clinical development of CT, a high-risk product, may have validity in a field requiring clinical benefit, an evaluation plan for the appropriate mechanism of action and efficacy should be established.
Conclusion and Suggestions
It is clear that CT products with more diverse origins and characteristics will be developed for a much wider range of clinical indications in the future. Responding to such a CT product development product through general quality and safety standards common to all molecules that have been applied to conventional products is impossible. Developing the ability to evaluate safety concerns and the value of each product justifying it based on an accurate understanding of the characteristics of the product is essential. Furthermore, the ability to generate, secure, and combine appropriate pieces of evidence to support the rationale of the development should be developed. Currently, human resources related to the development of CT products are mainly engaged in basic research, and the achievements are remarkable. However, ultimately, these CT products should be applied clinically and should be able to create practical added values. For this, training sufficient personnel with expertise related to the development of such products is necessary. Moreover, these experts should be evenly distributed not only to the sponsor but also to the regulatory agency to create a condition for rational discussion by pipeline. Through the experience accumulated in this process, the chance will be created for refining regulations, and a more effective development process of CT products will be possible.
 XML Download
XML Download