

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Staphylococcus aureus is one of the most frequent causes of hospital- and community-acquired infections worldwide. It is the leading cause of various infections, including skin and soft-tissue infections, pneumonia, osteomyelitis, endocarditis, and bacteremia [1]. A key challenge in controlling S. aureus infections has been the emergence and spread of strains resistant to multiple antibiotics, including penicillin and methicillin [2]. Methicillin-resistant S. aureus (MRSA) infections are typically treated with vancomycin; however, strains with various degrees of vancomycin resistance are becoming increasingly prevalent [3, 4].
S. aureus isolates exhibiting a vancomycin minimal inhibitory concentration (MIC) higher than 16 μg/mL are considered vancomycin-resistant S. aureus (VRSA). Resistance in VRSA is conferred by the vanA operon encoded on transposon Tn1546, originally a part of the vancomycin-resistant enterococci conjugative plasmid [5, 6]. S. aureus isolates with moderately increased vancomycin MIC values (4–8 μg/mL) are known as vancomycin-intermediate resistant S. aureus (VISA) [5]. The genetic basis of the VISA phenotype appears to be complex, likely involving multiple genes, and is less well understood than that of VRSA [7].
Whole-genome comparisons and candidate gene sequencing as well as studies of VISA phenotypic characteristics have identified numerous single-nucleotide polymorphisms (SNPs) in VISA strains compared to that in vancomycin-susceptible S. aureus (VSSA) strains [7-9]. Of the many SNPs associated with vancomycin resistance, the genes most frequently associated with the VISA phenotype are the regulatory genes walKR, vraRS, and graRS, as well as the gene encoding RNA polymerase subunit B (rpoB) [10, 11]. While these loci are common, the mutations detected within them are highly variable, and not all SNPs have a functional impact. Furthermore, very few SNPs have been experimentally validated to be responsible for the transition from VSSA to VISA.
In routine practice, the standard method for determining the vancomycin MIC is broth microdilution (BMD) or the E-test. These methods are time-consuming, labor-intensive, and error-prone, leading to a high degree of ambiguity in the results [12]. Importantly, failure to provide appropriate therapy promptly is associated with increased mortality [13]. Therefore, a suitable detection tool that can overcome the above-mentioned limitations and distinguish VISA from VSSA isolates needs to be developed.
We performed a microbial genome-wide association study (mGWAS) to identify SNPs that can discriminate between VISA and VSSA isolates and developed a TaqMan-based SNP genotyping assay for the detection of mutations associated with the VISA phenotype.
METHODS
Bacterial strains and phenotypes
The bacterial strains (N=26) used in this study are listed in Table 1. Clinical strains of S. aureus isolated from multiple body sites of patients (including blood, sputum, and wounds) between 2001 and 2011 were obtained from the VRSA surveillance system in Korea and analyzed retrospectively. Vancomycin MICs were determined using the standardized BMD, agar dilution, and E-test methods [12]. According to the CLSI definition [14], S. aureus isolates with vancomycin MICs of 4–8 μg/mL, as determined by at least one of the aforementioned methods, were classified as VISA. An E-test MIC value of 3 was considered equal to 4 μg/mL determined by BMD or agar dilution. S. aureus ATCC 29213 was used as a QC strain.
Table 1
Bacterial strains used in this study
Abbreviations: AD, agar dilution; BMD, broth microdilution; ET, E-test; MIC, minimal inhibitory concentration; ST, sequence type; SCCmec, staphylococcal cassette chromosome mec; spa, staphylococcal protein A; VSSA, vancomycin-susceptible Staphylococcus aureus; VISA, vancomycin-intermediate resistant S. aureus.
![]()
Whole-genome sequencing (WGS)
Genomic DNA was isolated from overnight bacterial cultures of the strains using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA), with the enzyme pretreatment step modified to 50 μg/mL lysostaphin and 500 μg/mL lysozyme in 10 mM Tris-HCl 1 mM EDTA (pH 8.0). Sequencing libraries were prepared using the TruSeq DNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. WGS was performed using the Illumina MiSeq platform (Illumina). The paired-end sequencing reads were assembled using CLC Genomics Workbench 7.3 (CLC Bio, Aarhus, Denmark). Genes were predicted using Glimmer 3 [15] and annotated by homology searches against the Clusters of Orthologous Groups and SEED databases (https://theseed.org) [16]. To validate the results, the assembled sequences were compared with the N315 and Mu50 MRSA reference genomes.
SNP calling and mGWAS
In total, 42 whole-genome sequences, including the sequences from the 26 clinical strains and 16 publicly available complete genomes (Supplemental Data Table S1), were used for mGWAS. Fig. 1 provides an overview of the bioinformatics workflow, which is compatible with WGS data. Before SNP calling, we applied a two-step QC process to clean up raw reads using PRINSEQ v.0.20.3 [17]. First, we removed reads with two or more ambiguous bases or a mean Phred quality score <20. Second, low-quality bases (Phred quality score ≤19) were trimmed from the 3´-end, and if after the trimming, the read length was <70 bp or the mean Phred quality score of the read was <20, it was discarded. The paired-end reads that fulfilled the above-mentioned quality criteria were mapped against the S. aureus N315 reference genome (GenBank accession: NC_002745) using Burrows–Wheeler Aligner for short-read alignment v.0.702 [18]. SNPs were called using SAMtools and BCFtools (https://www.htslib.org). The SNPs were functionally annotated using an in-house developed Perl script. We conducted an association study on a set of S. aureus whole-genome sequences to identify SNPs that were significantly associated with VISA using the PLINK software package (https://zzz.bwh.harvard.edu/plink/) [19]. QC of the identified SNPs was performed using the PLINK options -geno 0.9 and -maf 0.05.
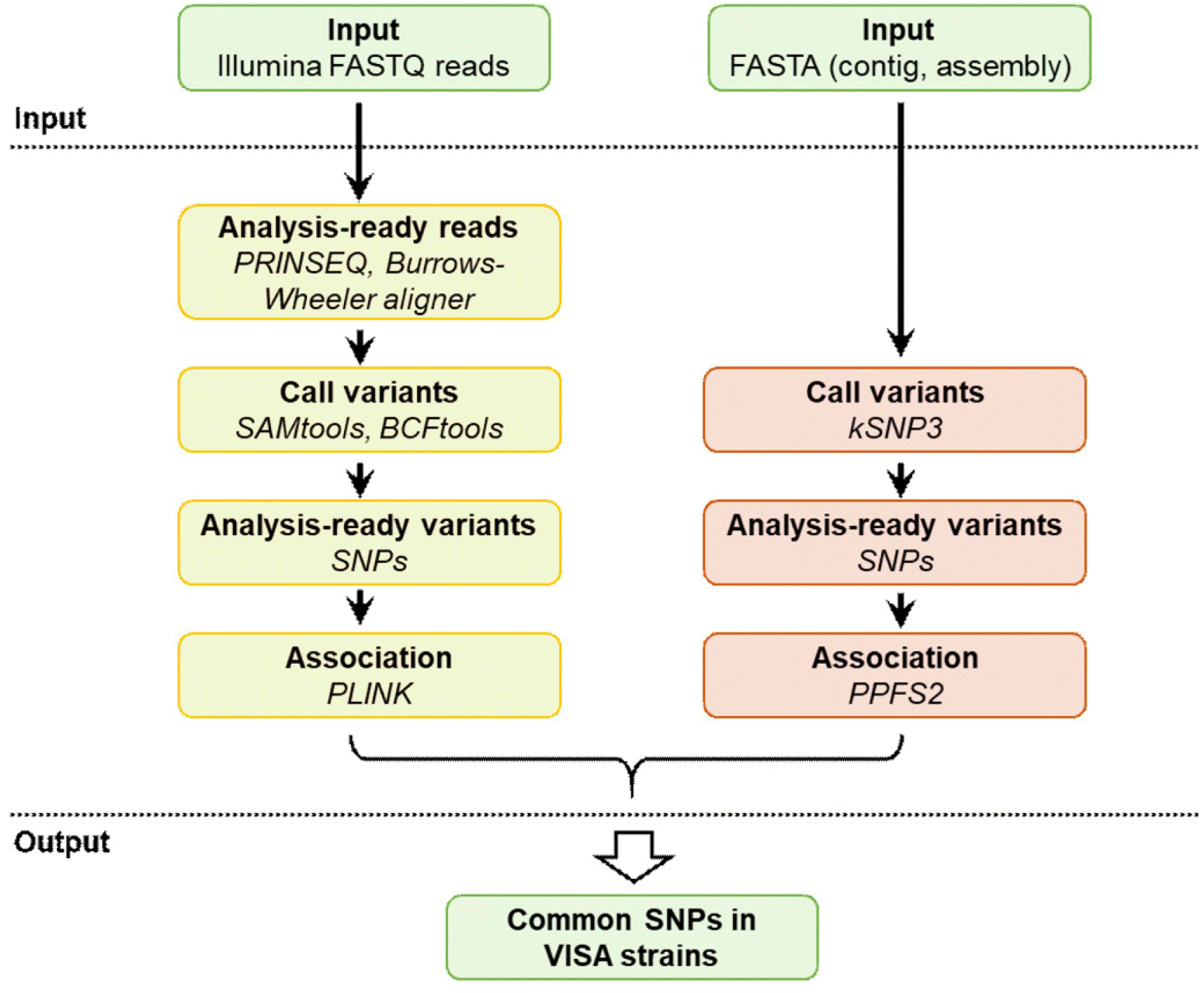
In addition, we used the Predict Phenotypes From SNPs (PPFS) package add-on to kSNP3.0, a program that can identify SNPs in a data set of hundreds of microbial genomes [20, 21]. The PPFS package consists of five programs: PPFS, PickPhenotypeSubset, GetSNPprobes, DiagnosticSNPs, and causal SNPs. Association analysis was performed using the DiagnosticSNPs program. To predict the phenotype, we constructed a.pheno file in which the phenotype of each genome was listed as VISA (1), VSSA (0), or unknown (?). By comparing the known with the predicted phenotypes, the numbers of true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN) were determined. The accuracy of the analysis was determined as (TP+TN)/(TP+FP+TN+FN). The positive predictive value was calculated as TP/(TP+FP), whereas the negative predictive value was calculated as TN/(TN+FN). The SNPs obtained from the mGWAS were validated in two steps: (i) Basic Local Alignment Search Tool (BLAST) analysis, and (ii) PCR and sequencing of clinical VSSA and VISA strains other than those used for WGS (Supplemental Data Tables S2 and S3).
TaqMan SNP genotyping assay development and assessment
The TaqMan assay was designed using a set of pre-validated SNPs and was performed in 20-μL reactions in 96-well plates, using low ROX as a passive reference dye. Allelic specificity of the TaqMan assay was ensured by using two probes, one labeled with FAM dye and the other with VIC dye (Supplemental Data Table S4). Genotyping was performed on an Applied Biosystems 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using AB 7500 software v.2.0.6 following the default standard allelic discrimination genotyping assay protocol. The assay was carried out in a single tube in a 20-μL reaction volume containing 10 μL of 2× master mix, 5 μL of each primer and probe mix, and 5 μL of DNA. The thermal cycles were as follows: a pre-read stage at 60°C for 1 minute, initial denaturation at 95°C for 10 minutes, and 40 cycles of denaturation at 95°C for 15 seconds and annealing at 60°C for 1 minute, and a post-read stage at 60°C for 1 minute. Analytical sensitivity was assessed using DNA input amounts ranging from 100 to 0.1 ng DNA per reaction. The limit of detection (LOD) of the assay was evaluated using the normalized reporter signal Rn.
RESULTS
Loci associated with reduced vancomycin susceptibility
Using the core genome of N315 (E-test MIC, 0.5 μg/mL) as the reference for SNP calling, we identified 47 SNPs in the 26 strains sequenced in this study. We used PLINK to identify SNPs associated with vancomycin-intermediate resistance within this set. All 47 SNPs were found to be associated with the VISA phenotype. As PLINK is frequently used for haploid eukaryotic GWAS, we used the kSNP3.0 and PPFS packages for SNP calling and mGWAS. kSNP3.0 identifies SNPs in bacterial and viral genomes (finished genomes, genome assemblies, or raw reads) without using genome alignment or reference genomes. Using kSNP3.0 and PPFS, we obtained 46 VISA phenotype-associated SNPs.
Forty-six SNPs were consistent between the PLINK and PPFS results (Table 2). Of these SNPs, 27 were nonsynonymous, resulting in amino-acid substitution, whereas 10 were synonymous. Nine SNPs were located in intergenic regions (Table 2). The proportion of these 46 SNPs in the 28 VISA strains studied ranged from 78.6% to 85.7%, whereas the SNPs were not identified in the 14 VSSA strains (Table 2). All VISA strains harboring SNPs belonged to the ST5 clonal lineage. To confirm nonsynonymous SNPs associated with the VISA phenotype, the 27 nonsynonymous SNPs were validated using BLAST analysis and PCR and sequencing. Based on the BLAST results of strains with known phenotypes among genomes registered in National Center for Biotechnology Information, 12 SNPs related to the VISA phenotype were selected from the 27 nonsynonymous SNPs. In the second step, the association of the SNPs with the VISA phenotype was tested in 18 VSSA and 26 VISA clinical strains using PCR and sequencing. Finally, four SNPs found only in VISA strains and not in VSSA strains were selected: SA_RS01235 (G203S), SA_RS09725 (V171A), SA_RS12250 (I48F), and SA_RS12550 (G478A).
Table 2
The 46 SNPs associated with the VISA phenotype
| N315 locus tag* | Position | Mutation | Amino-acid change† | Biological function | Proportion of SNPs in VISA (%) |
|---|---|---|---|---|---|
| SA_RS01235 → | 249,622 | G→A | G203S | Gfo/Idh/MocA family oxidoreductase | 85.7 |
| SA_RS01705 ← / → SA_RS01710 | 350,179 | A→C | Intergenic (–335/–249) | Branched-chain amino acid transporter II carrier Protein/5´-nucleotidase | 78.6 |
| SA_RS01765 ← | 362,625 | G→C | S55T | RpiR family transcriptional regulator | 78.6 |
| SA_RS01845 ← | 378,175 | C→A | P530H | Hypothetical protein | 85.7 |
| SA_RS01985 ← / → SA_RS01990 | 407,772 | A→C | Intergenic (–302/–360) | Cystathionine gamma synthase/chromosome-partitioning protein ParB | 78.6 |
| SA_RS02445 → | 493,702 | A→G | A1013A | Glutamate synthase | 82.1 |
| SA_RS02450 → | 495,666 | A→G | P162P | Glutamate synthase subunit beta | 85.7 |
| SA_RS02640 → | 528,901 | G→A | E112E | Ribose phosphate pyrophosphokinase | 78.6 |
| SA_RS02980 → | 596,506 | T→C | A137A | Ribulokinase | 85.7 |
| SA_RS03020 → / → SA_RS03025 | 605,117 | C→T | Intergenic (+331/–98) | FMN-dependent NADPH-azoreductase/serine aspartate repeat-containing protein C | 78.6 |
| SA_RS03830 → / → SA_RS03835 | 765,823 | A→T | Intergenic (+57/–417) | Allophanate hydrolase/lipoteichoic acid synthase | 78.6 |
| SA_RS03930 → | 788,363 | T→C | G122G | Iron ABC transporter permease | 85.7 |
| SA_RS04890 → | 978,290 | C→T | S36L | GTP pyrophosphokinase | 78.6 |
| SA_RS04980 → | 1,001,999 | A→T | M354L | 2´,3´-Cyclic nucleotide 2´-phosphodiesterase | 85.7 |
| SA_RS05400 → | 1,081,200 | A→T | E163V | Chitinase | 78.6 |
| SA_RS05685 → / → SA_RS05690 | 1,135,543 | G→A | Intergenic (+41/–219) | Membrane protein/fibrinogen-binding protein | 78.6 |
| glpK → | 1,298,748 | G→A | E371K | Glycerol kinase | 78.6 |
| SA_RS06580 → | 1,320,949 | G→A | D189N | LuxR family transcriptional regulator | 78.6 |
| ebh ← | 1,446,514 | T→C | H3852H | Extracellular matrix-binding protein | 85.7 |
| SA_RS07360 ← / ← SA_RS07365 | 1,507,988 | G→A | Intergenic (–62/+30) | Nucleoside diphosphate kinase/heptaprenyl diphosphate synthase subunit II | 78.6 |
| SA_RS07605 ← | 1,551,898 | G→A | R24H | 6-Phosphogluconate dehydrogenase decarboxylating | 85.7 |
| SA_RS07790 ← | 1,582,579 | C→T | G194G | glucokinase | 78.6 |
| dnaK ← | 1,615,059 | C→T | N96N | Molecular chaperone DnaK | 78.6 |
| SA_RS08080 ← | 1,636,342 | T→C | I63T | Iron transporter | 85.7 |
| SA_RS08520 ← | 1,723,724 | A→T | K335I | DNA polymerase I | 78.6 |
| SA_RS08560 ← | 1,734,746 | A→G | G423G | Pyruvate kinase | 78.6 |
| SA_RS08765 ← | 1,781,936 | G→A | G56R | Acetyl-CoA synthetase | 85.7 |
| SA_RS08780 ← | 1,784,869 | A→T | E105D | Catabolite control protein A | 78.6 |
| SA_RS08975 ← | 1,833,017 | C→T | R119C | Autolysin | 85.7 |
| SA_RS09210 ← | 1,870,103 | C→T | T43I | Leucotoxin LukDv | 78.6 |
| SA_RS09725 ← | 1,942,181 | T→C | V171A | Acyl-CoA hydrolase | 85.7 |
| SA_RS09905 → / ← SA_RS09910 | 1,977,756 | C→A | Intergenic (+112/+180) | Staphostatin A/hypothetical protein | 78.6 |
| SA_RS10425 → | 2,055,468 | T→A | L212M | Potassium transporter KtrB | 85.7 |
| SA_RS10915 ← | 2,149,914 | G→A | E20K | Aminopyrimidine aminohydrolase | 78.6 |
| SA_RS11170 ← | 2,194,608 | C→T | E295K | Mannose 6 phosphate isomerase | 85.7 |
| SA_RS11400 ← | 2,248,253 | A→T | T233T | ABC transporter substrate-binding protein | 78.6 |
| SA_RS11550 → | 2,278,530 | A→G | T92A | Toxin | 78.6 |
| SA_RS11870 ← | 2,327,335 | A→G | K114E | Cyclic pyranopterin monophosphate synthase | 85.7 |
| SA_RS12195 ← | 2,386,988 | C→A | P352Q | Urocanate hydratase | 85.7 |
| SA_RS12200 → | 2,388,608 | T→C | L136S | LysR family transcriptional regulator | 85.7 |
| SA_RS12250 ← | 2,397,878 | A→T | I48F | Sodium ABC transporter ATP-binding protein | 85.7 |
| SA_RS12550 ← | 2,458,970 | G→C | G478A | Nitrite reductase | 85.7 |
| SA_RS13145 ← / ← SA_RS13150 | 2,575,425 | C→A | Intergenic (–85/+49) | Hypothetical protein/gluconate permease | 78.6 |
| SA_RS13485 ← / ← SA_RS13490 | 2,646,865 | A→T | Intergenic (–16/+497) | O-acetyltransferase OatA/GNAT family acetyltransferase | 78.6 |
| SA_RS13735 ← | 2,689,707 | C→T | P294L | Malate:quinone oxidoreductase | 78.6 |
| SA_RS13995 ← | 2,748,292 | G→T | R524S | Accessory Sec system translocase SecA2 | 78.6 |
Abbreviations: SNP, single-nucleotide polymorphism; VISA, vancomycin-intermediate resistant Staphylococcus aureus; FMN, flavin mononucletotide; ABC, ATP-binding cassette; ATP, adenosine triphosphate; GTP, guanosine triphosphate; GNAT, Gnc5-related N-acetyltransferase; SecA2, protein translocase subunit SecA2.
![]()
Rapid detection of SNPs associated with reduced vancomycin susceptibility
We employed a TaqMan SNP genotyping assay that discriminates between VISA and VSSA. Allele-specific primers and probes were designed for the four selected SNPs. The LOD for SNP genotyping was determined using a dilution series of input genomic DNA ranging from 100 to 0.1 ng DNA per reaction. The assay successfully detected all SNPs using as little as 0.2 ng DNA per reaction (Supplemental Data Tables S5 and S6). Out of the 44 clinical strains (Supplemental Data Table S2), 20 randomly selected strains (10 VSSA and 10 VISA) were examined to verify that the SNP genotyping assay accurately discriminated between VISA and VSSA. Allelic discrimination plots for all four SNPs showed good discrimination between the VISA and VSSA phenotypes (Fig. 2).
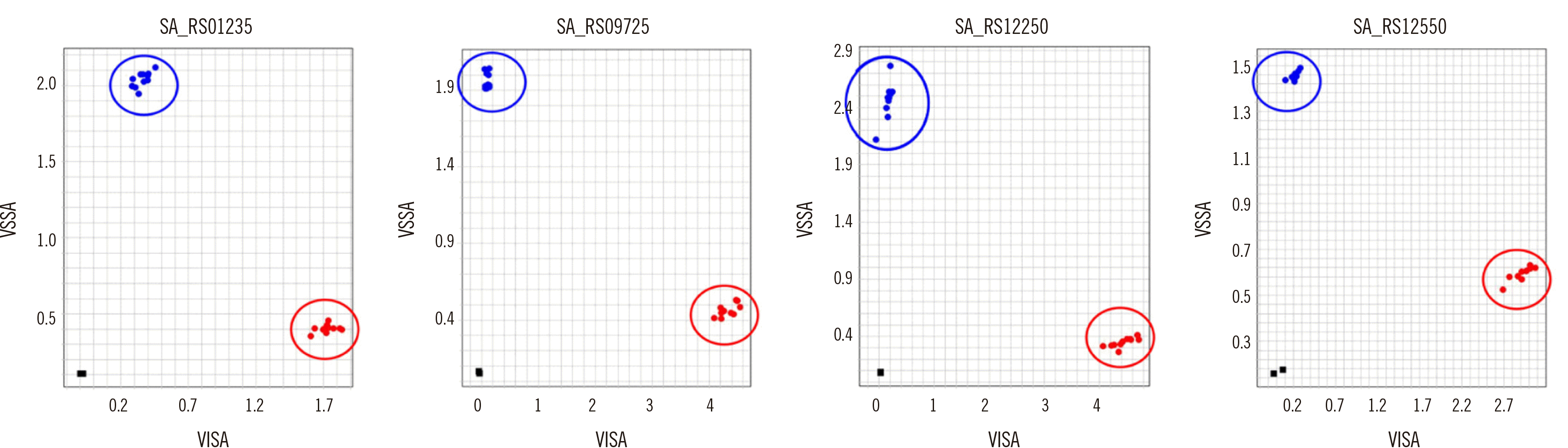
Fig. 2
Allelic discrimination plots for the four SNPs using the TaqMan genotyping assay on 20 samples (10 VSSA and 10 VISA). Red (VISA) and blue (VSSA) dots represent the homozygous genotypes. The square at the bottom left of the plot is the no-template control.
Abbreviations: SNP, single-nucleotide polymorphism; VISA, vancomycin-intermediate resistant Staphylococcus aureus; VSSA, vancomycin-susceptible S. aureus.
![]()
DISCUSSION
SNPs are the most widely used molecular markers owing to their genome-wide distribution and low cost of detection compared to other markers [22]. PCR-based allelic discrimination methods have broad applications in SNP detection in genetics and genomics. The TaqMan SNP genotyping assay developed in this study successfully identified four SNPs selected from the mGWAS results: SA_RS01235 (G203S), SA_RS09725 (V171A), SA_RS12250 (I48F), and SA_RS12550 (G478A).
mGWAS is a new research area aimed at identifying genetic variants in microbial genomes that are associated with host or microbe phenotypes, such as carriage in humans and virulence in microbes [23, 24]. mGWAS has been crucial in the identification of novel genomic markers of drug resistance. Alam, et al. [25] used a GWAS to identify mutations in the RNA polymerase rpoB gene of S. aureus that were significantly associated with the clinically important VISA phenotype. Farhat, et al. [26] studied 116 Mycobacterium tuberculosis strains and found evidence of positive selection in 39 genomic regions in resistant isolates. mGWAS has been used to detect genes and genetic variants associated with resistance to 17 antimicrobials in 3,144 isolates from four taxonomically diverse and recombining bacterial species [27]. Understanding the genetic architecture of a particular drug resistance phenotype allows exploring other genetically correlated phenotypes and informing treatment, drug design, and repositioning decisions.
Two-component regulatory systems, including the walKR, vraSR, and graSR genes, have been linked to reduced vancomycin susceptibility [7, 10, 11, 13]. These regulatory genes are involved in cell wall biosynthesis and degradation and the stress-responsive alternative sigma factor σB [7]. Another mechanism by which S. aureus acquires vancomycin-intermediate resistance is by modulating its physiology and metabolism to permit growth in the presence of vancomycin, known as adaptive resistance [28]. The metabolic adaptations include increased acetogenesis, carbon flow through the pentose phosphate pathway, wall teichoic acid and peptidoglycan precursor biosynthesis, and purine biosynthesis and decreased tricarboxylic acid cycle activity. As the metabolic adaptations involve central metabolism, it is likely that a broader array of metabolic changes are involved in vancomycin-intermediate resistance [29]. However, information on the contributions of most mutations in the regulatory genes to metabolic changes is lacking [28–30].
We identified four novel SNPs that distinguish VSSA from VISA—SA_RS01235 (G203S), SA_RS09725 (V171A), SA_RS12250 (I48F), and SA_RS12550 (G478A)—encoding Gfo/Idh/MocA family oxidoreductase, acyl-CoA thioesterase, ABC transporter ATP-binding protein, and NAD(P)/FAO-dependent oxidoreductase (nitrate reductase), respectively. These genes are associated with general metabolic pathways, including lipid metabolism, nitrogen assimilation, and sodium transport.
A recent systematic review revealed that the most epidemic genotypes of heterogeneous VISA/VISA are ST5 and ST239, which are predominant among hospital-associated MRSA strains [31]. Consistent herewith, an epidemiological study in Korea showed that ST5 and ST239 strains are predominant in hospital-associated infections and mainly include heterogeneous VISA/VISA strains [32]. Although we analyzed a few minor genotypes, the majority of strains used in this study belonged to ST5 (24/26, 92%). Furthermore, the four novel SNPs associated with reduced vancomycin susceptibility were observed only in the ST5 lineage. The finding of VISA-associated mutations at a genome-wide significance level in ST5 strains indicates that vancomycin-intermediate resistance occurred through convergent evolution rather than random genetic rearrangement. Park, et al. [32] demonstrated that accumulated genetic variants are involved in reduced vancomycin susceptibility through different mechanisms in each clonal lineage. Further genome sequencing studies on larger collections of VISA strains with varied genetic backgrounds are needed to determine the gene variants that cause vancomycin-intermediate resistance.
We employed a SNP assay that discriminated between VISA and VSSA. TaqMan-based allelic discrimination offers high-throughput analysis and accurate SNP detection. The TaqMan assay successfully detected all SNPs using as little as 0.2 ng DNA per reaction. Classic gene sequencing methods, such as Sanger sequencing, are time-consuming and not appropriate for studies involving large populations and small SNP numbers. This fluorescence-based procedure considerably simplifies the assay protocol by eliminating the need for gel electrophoresis and visual assessment of bands. Thus, our assay shortens the sample turnaround time in the clinical laboratory. The total process time from culture sampling to result was less than 4 hours.
Our study had several limitations. We found four novel SNPs significantly associated with reduced vancomycin susceptibility; however, gene-knockout and -overexpression studies and studies in other VISA lineages are needed to validate that these genes are determinants of vancomycin resistance. Further, the number of well-characterized clinically isolated VISA strains in this study was limited, whereas mGWAS requires large, complex sample collections. A large collection not only increases the statistical strength of association studies but also allows investigating lineage-specific traits and/or low-frequency variants.
In conclusion, we developed a high-throughput TaqMan SNP genotyping assay that allows rapid detection of VISA by discriminating VISA and VSSA. This assay may be utilized to accelerate therapeutic decision making and allow earlier and more appropriate antimicrobial treatment highly pertinent to vancomycin resistance. In future, this assay may help decrease the spread of resistant strains by avoiding the use of unnecessary antimicrobial agents.
 XML Download
XML Download