

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
AML is a biologically heterogeneous disease, resulting in varying clinical outcomes [1]. Although most AML patients achieve cytomorphologic remission, recurrence is reported in approximately 50% of patients who achieved remission after initial treatment [2, 3]. In light of the high frequency of recurrence, the current criterion for gross remission of bone marrow, i.e., a residual tumor burden (measurable residual disease, MRD) <5%, has been questioned [4]. Recently developed detection methods have enabled the detection of a low leukemic burden of 10–3–10–6 cells [4, 5]. A more sensitive MRD detection method would allow for a more reliable assessment of remission status than microscopic examination of bone marrow smears. MRD analysis results may help refine disease prognosis and enable early intervention, if necessary, and may guide the use of customized therapy for patients [4, 6]. Post-transplant monitoring is another application of MRD analyses.
There is significant genetic heterogeneity among AML patients and within individual patients [7]. Founding genetic mutations exist before the progression of AML, and as the disease progresses, various subclones are formed. The mutations disappear or expand due to the selective pressure induced by chemotherapy treatment, resulting in a wide mutation spectrum [8, 9]. Genetic mutations in AML patients comprise various combinations of gene rearrangements as well as point mutations. Detecting these genetic mutations may aid in monitoring micro-existence cancers. However, as there are numerous genetic variants in AML, it is critical to determine which are clinically significant and can be detected with high sensitivity.
The European LeukemiaNet (ELN) recently proposed consensus guidelines for MRD monitoring [10]. The ELN MRD Working Party concluded that the risk of recurrence is high when MRD is detected through various test methods and recommended that MRD monitoring results be reflected in patient treatment plans [10].
Next-generation sequencing (NGS) can simultaneously detect multiple mutations, allowing easy identification of genetic heterogeneity. We designed a sensitive error-corrected targeted NGS panel for MRD that can be applied to routine clinical testing of AML patients.
MATERIALS AND METHODS
Patients and sample preparation
Twenty-three AML patients diagnosed at Yonsei University Severance Hospital, Seoul, Korea, between January 2018 and March 2020 were included in this study. We retrospectively performed NGS on 54 cryopreserved bone marrow or peripheral blood samples stored at –80°C from the 23 AML patients. The samples were subjected to NGS using a customized panel targeting 497 genes at leukemia diagnosis. Genomic DNA (gDNA) was extracted from samples using a QIAsymphony DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s guidelines. To reduce false-positive variants, gDNA was treated with uracil-DNA-glycosylase (UDG; NEB, Ipswich, MA, USA) per the manufacturer’s protocol [11]. Clinical information was obtained from electronic medical records. This study was approved by the Institutional Review Board (4-2021-0009) of Severance Hospital. Informed consent was waived for this study because participants had already consented to another study for use of their samples and data for secondary research.
Custom gene panel design
We selected 24 genes for the NGS-MRD panel based on information on diagnostic panel testing stored in our institution’s internal database in the last 4 years (Supplemental Data Table S1). Based on the prevalence of variants in 521 patients (346 AML and 175 ALL patients) who underwent gene panel testing at the time of diagnosis of acute leukemia in our institution, our MRD panel was expected to enable monitoring approximately 78.0% of AML and 37.1% of ALL patients, respectively (unpublished data). In the case of ALL, many patients only showed gene copy number alterations (CNAs; deletion/duplication), but we excluded CNA from the NGS-MRD panel because the detection sensitivity of NGS for CNAs is generally lower than that for single-nucleotide variants (SNVs) [12]. All coding exons and flanking intronic regions were included as target regions. Sets of double-stranded DNA probes, approximately 120 bp in length, were designed to hybridize to regions of interest and were synthesized by Dxome (Seoul, Korea). The total size of the capture region was estimated to be 87 kb.
Validation of the designed panel
To assess SNV detection performance, Horizon Tru-Q DNA reference material (Horizon Discovery, Cambridge, UK) was used. Tru-Q4 contains six validated somatic mutations targeted by the panel. Serial dilutions of the reference material were prepared using Horizon Tru-Q0 wild-type DNA as a diluent to generate mixtures with 5.00%, 0.50%, 0.250%, 0.125%, and 0.006% variant allele frequency (VAF). Droplet digital PCR (ddPCR) testing was performed on certain SNVs to verify the accuracy of the panel tests. Two SNVs were selected from the manufacturer’s commercial pre-designed and validated probes. Detailed ddPCR methods are provided in the Supplemental Materials and Methods.
NGS
Details on NGS library preparation are provided in the Supplemental Materials and Methods. Briefly, approximately 300 ng of gDNA was used to prepare sequencing libraries using a Twist Library Preparation EF Kit (Twist Bioscience, San Francisco, CA, USA). Target enrichment was performed using a capture probe, and sequencing was conducted on a NovaSeq 6000 instrument (Illumina, San Diego, CA, USA), achieving approximately 150 million reads per sample. Sequencing was performed using a 151-bp, dual-indexed, paired-end sequencing configuration.
Data analysis and variant calling
Trimmomatic was used for FASTQ data QC [13]. Reads from trimmed FASTQ data were aligned to the reference genome GRCh37/hg19 using Burrows–Wheeler Aligner (BWA-mem; v0.7.12) [14], and variants were identified using the PiSeq algorithm (Dxome), which was developed to verify the accuracy of molecular barcoding by calculating genome positions of mapped reads [15]. Variant annotation was performed using DxSeq software (Dxome), and annotated variants were classified into four tiers according to the Standards and Guidelines of the Association for Molecular Pathology [16]. All mutations were verified manually using Integrative Genomic Viewer [17].
Statistical analysis and data visualization
All statistical analyses were performed using Microsoft Excel (Microsoft Corp., Redmond, WA, USA) and MedCalc v18.2.1 (MedCalc Software, Ostend, Belgium). Correlations between variables were evaluated using the Passing–Bablok model and Pearson’s correlation coefficient (R). To compare of VAF differences between disease statuses, we used the Mann–Whitney U-test. Progression-free survival (PFS) was measured from the date of initial sampling before chemotherapy initiation to the date of progression, death from any cause, or last follow-up. Overall survival (OS) was measured from the date of initial sampling before chemotherapy initiation to the date of death or last follow-up. Survival analyses were performed using the Kaplan–Meier method. Statistical significance was defined as P<0.05.
RESULTS
Technical validation and run metrics
To optimize the amount of gDNA, number of PCR cycles, and adapter concentrations for use in high-sensitivity NGS-MRD tests in a genetic diagnostic setting, 100–300 ng of reference material was evaluated using the PiSeq algorithm QC parameters (Supplemental Data Table S2), focusing on sensitivity. The final test conditions were as follows: 200 ng of gDNA treated with UDG, six cycles for gDNA library amplification, and 15 cycles for target enrichment. The average sequencing depth for the patient samples was 78,931×, and the average on-target read percentage was 60.08%.
Validation of the NGS-MRD panel using reference material
Triplicates of serial dilutions of reference material (5.00%, 0.50%, 0.250%, 0.125%, and 0.006%) were analyzed to evaluate the linearity of the designed NGS panel (Fig. 1 and Supplemental Data Table S3). The dilution test showed excellent linearity and a strong correlation between expected and observed clonal frequencies (R>0.99). The average frequencies of KRAS c.35G>A and NRAS c.182A>G detected by the NGS panel and by ddPCR are presented in Fig. 2. The NGS test reproducibly detected all six tested mutations in the three dilution series samples with a sensitivity of 0.25%. Our test showed excellent linearity, high concordance with ddPCR, and high sensitivity.
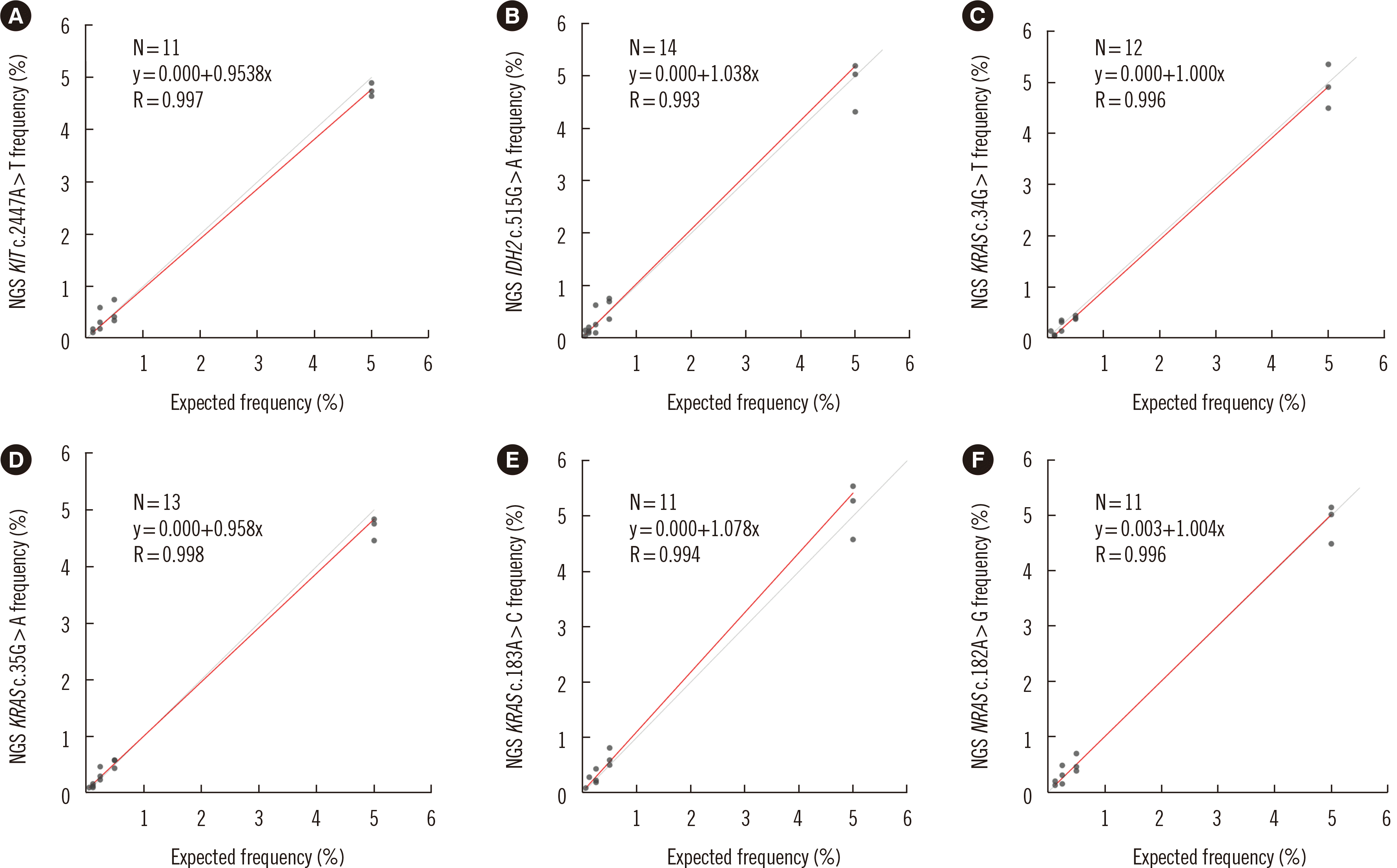
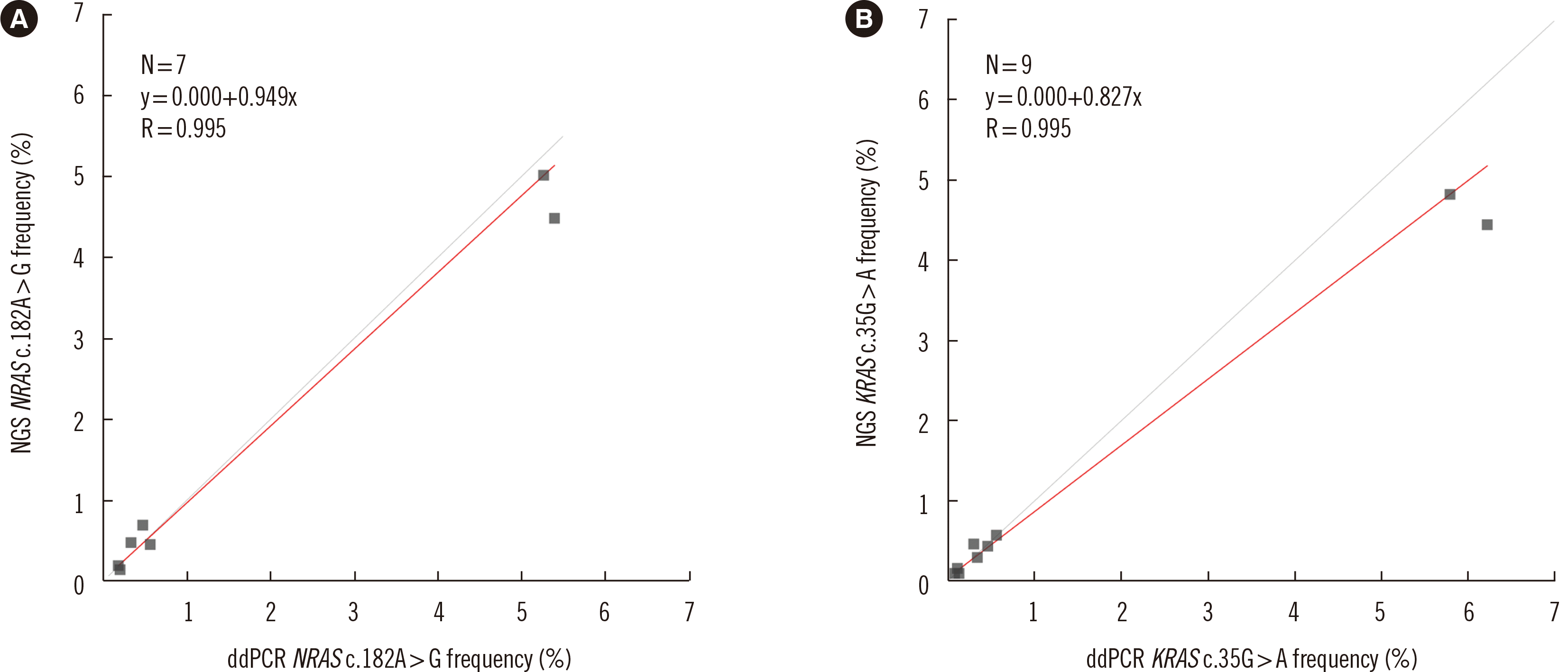
Fig. 1
Linearity range and correlation of mutant allele measurement between gravimetric dilution and NGS-MRD tests. (A) KIT c.2447A>T, (B) IDH2 c.515G>A, (C) KRAS c.34G>T, (D) KRAS c.35G>A, (E) KRAS c.183A>C, and (F) NRAS c.182A>G. The results of the linearity analyses using serially diluted reference material ranged from 5.00% to 0.06%. The best-fit regression equation and correlation coefficient (R) for each variant are indicated in the plots.
Abbreviations: NGS, next-generation sequencing; MRD, measurable residual disease.
![]()
Fig. 2
Comparative evaluation of the NGS-MRD and ddPCR tests. The results of the linearity analyses using serially diluted reference material ranged from 5.00% to 0.06%. The best-fit regression equation for (A) the NRAS c.182A>G mutation was y=0.949x, and that for (B) the KRAS c.35G>A mutation was y=0.827x.
Abbreviations: NGS, next-generation sequencing; MRD, measurable residual disease; ddPCR, droplet digital PCR.
![]()
Association of NGS-MRD panel results with clinical characteristics and outcomes
Patient clinical and biological characteristics are presented in Table 1. The majority of the patients (18/23, 78.3%) received hematopoietic stem cell transplantation (HSCT), and 78.3% harbored no gene rearrangements related to AML. The variants and their detection frequencies at diagnosis and after one month of chemotherapy (M1) in the 23 patients are provided in Supplemental Data Table S4. Most of the patients reached morphologic complete remission (CR) after induction of chemotherapy (22/23, 95.7%), but the NGS-MRD panel detected mutations at the time of diagnosis in 13 patients (13/22, 59.1%). Among these 13 patients, three harbored variants only in DNMT3A (0.57% for patient #3, 21.32% for patient #21) or ASXL1 (5.34%, patient #23); the presence of variants in these genes should be carefully interpreted because they are age-related clonal hematopoiesis genes. The median VAF in serial follow-up samples differed significantly between morphologic remission samples (bone marrow blasts <5%) (0.00%, interquartile range [IQR], 0.00%–0.13%) and MRD-positive samples (bone marrow blasts >5%) (39.73%, IQR, 0.00%–46.10%) (P<0.001, Mann–Whitney U-test) (Fig. 3, Supplemental Data Table S5). With respect to the sample type, only the median VAFs for the serial bone marrow follow-up samples differed significantly between the two groups (P<0.001). In CR samples, mutations were most frequently detected in DNMT3A (N=14, range 0.15%–30.21%) and NPM1 (N=7, range 0.12%–0.79%) (Supplemental Data Fig. S1).
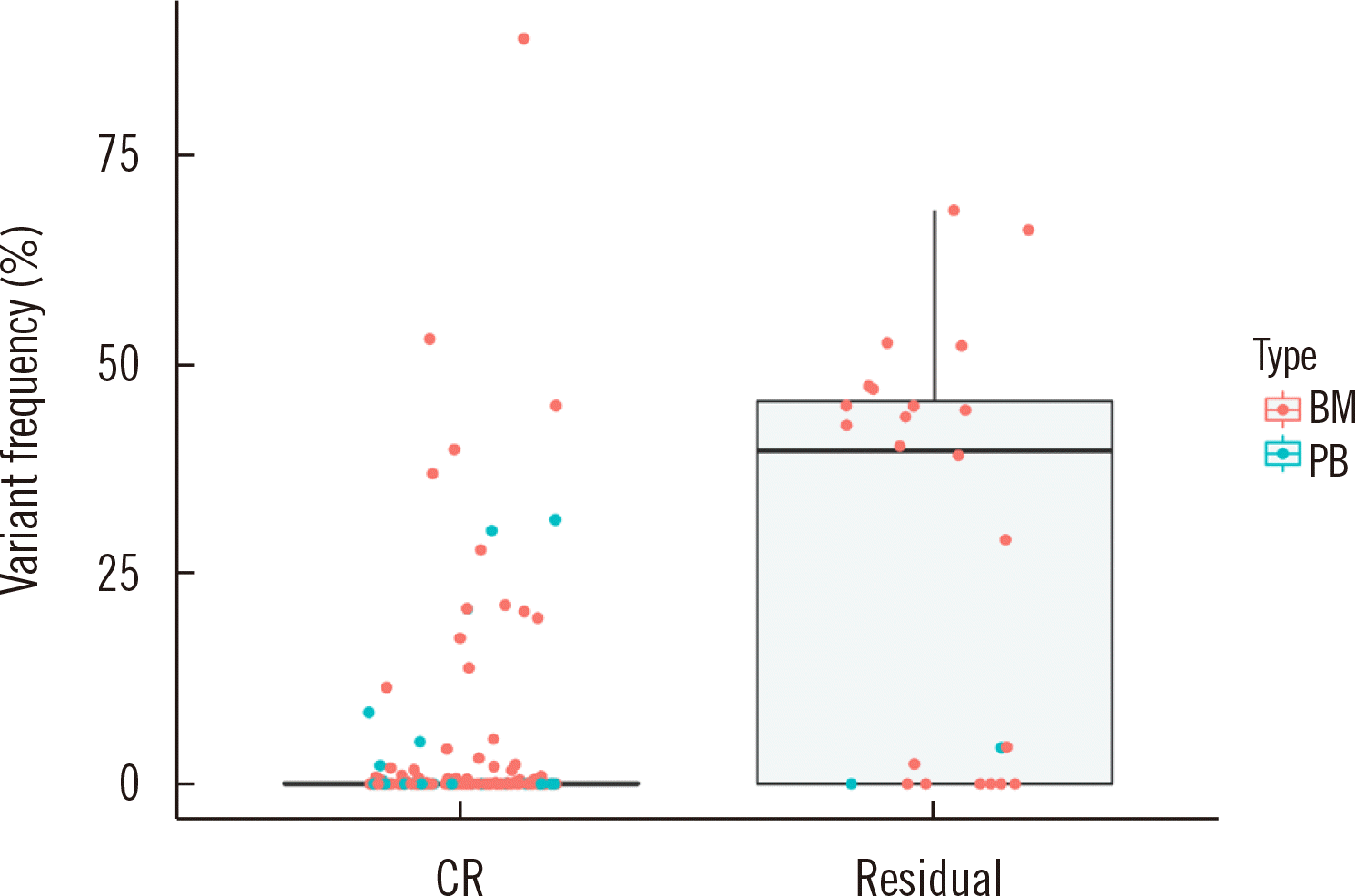
Fig. 3
Comparison of the variant frequency distribution in samples from patients in complete remission and those with residual disease. The median variant frequency of complete remission samples was 0.000%, whereas that of residual disease was 39.731%.
Abbreviations: CR, complete remission; BM, bone marrow sample; PB, peripheral blood sample.
![]()
Table 1
Patient characteristics (N=23)
| Characteristic | N (%) |
|---|---|
| Male sex | 9 (39.1) |
| Mean age (yr) | 47.4 |
| HSCT | |
| Yes | 18 (78.3) |
| No | 5 (21.7) |
| Survival | |
| Survive | 11 (47.8) |
| Death | 9 (39.1) |
| Follow-up loss | 3 (13.0) |
| Rearrangement* | |
| CBFB-MYH11 | 2 (8.7) |
| P2RY8-CRLF2 | 1 (4.3) |
| KMT2A-SEPT5 | 1 (4.3) |
| RUNX1-RUNX1T1 | 1 (4.3) |
| Not detected | 18 (78.3) |
![]()
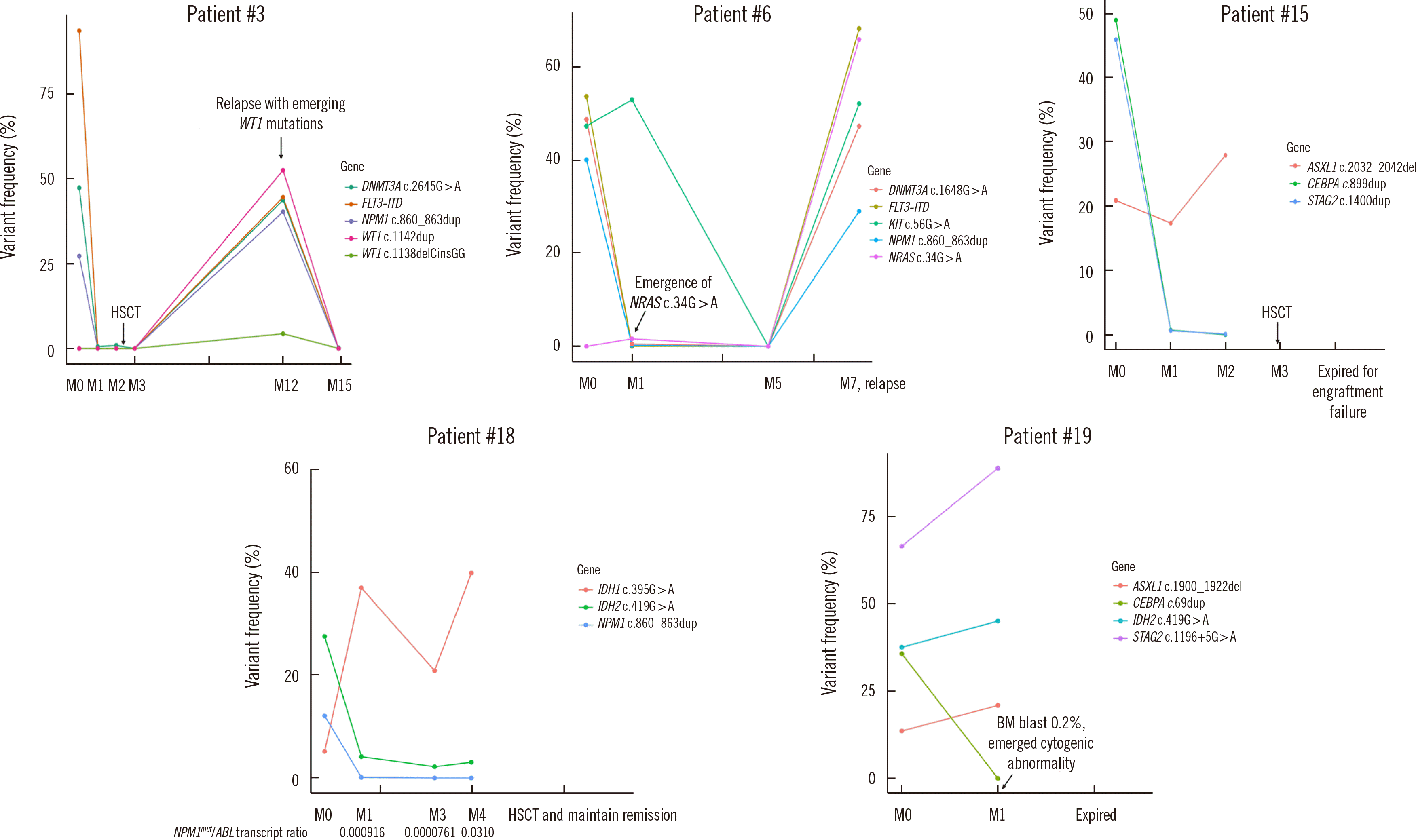
Representative serial MRD profiles for five patients are presented in Fig. 4. Newly emerging clones in relapsed patient samples were identified in patients #3 and #6. Patient #3 was initially diagnosed as having AML with mutated NPM1; DNMT3A c.2645G>A with FLT3-internal tandem duplication (ITD) mutations were also detected. After induction of chemotherapy, the NPM1 and FLT3-ITD mutations were cleared, whereas DNMT3A c.2645G>A (1.61%) was present at a very low allele frequency (<1.0%) in morphologic remission samples. When applying the NGS-MRD test to the sample after HSCT, the mutations identified initially were not detected. Nine months after HSCT, the patient relapsed, and a WT1 gene mutation that had not been present at the time of diagnosis was detected. Patient #6 showed morphologic remission after initial chemotherapy (bone marrow blasts, 0.8%) but residual KIT c.56G>A at a high allele frequency together with a newly emerged NRAS c.34G>A mutation (1.61%). The patient eventually relapsed, and the NRAS c.34G>A mutation identified in the M1 sample was one of the major mutations detected at relapse. Patients #15, #18, and #19 had discrepant bone marrow morphology and NGS-MRD results. Patient #15 had consistent high-frequency ASXL1 residual mutations and later died due to engraftment failure. Patient #18 showed clearance of only NPM1 on the NGS-MRD panel, but quantitative PCR showed an NPM1mut/ABL1 transcript ratio of 0.0000761 in the three-month sample. However, in the four-month sample, there was a discrepancy between the NGS-MRD and NPM1 quantitative PCR results (0.0% and transcript ratio of 0.031, respectively). Patient #19 was diagnosed as having AML with myelodysplasia-related change and showed clearance of the CEBPA mutation in the M1 sample. The percentage of bone marrow blasts declined to 0.2%, but dysplasia was still present and an emergent cytogenetic abnormality was detected (46,XY,?der(14)t(11;14)(q13;q32)[2]/46,XY[38]) in the M1 sample.
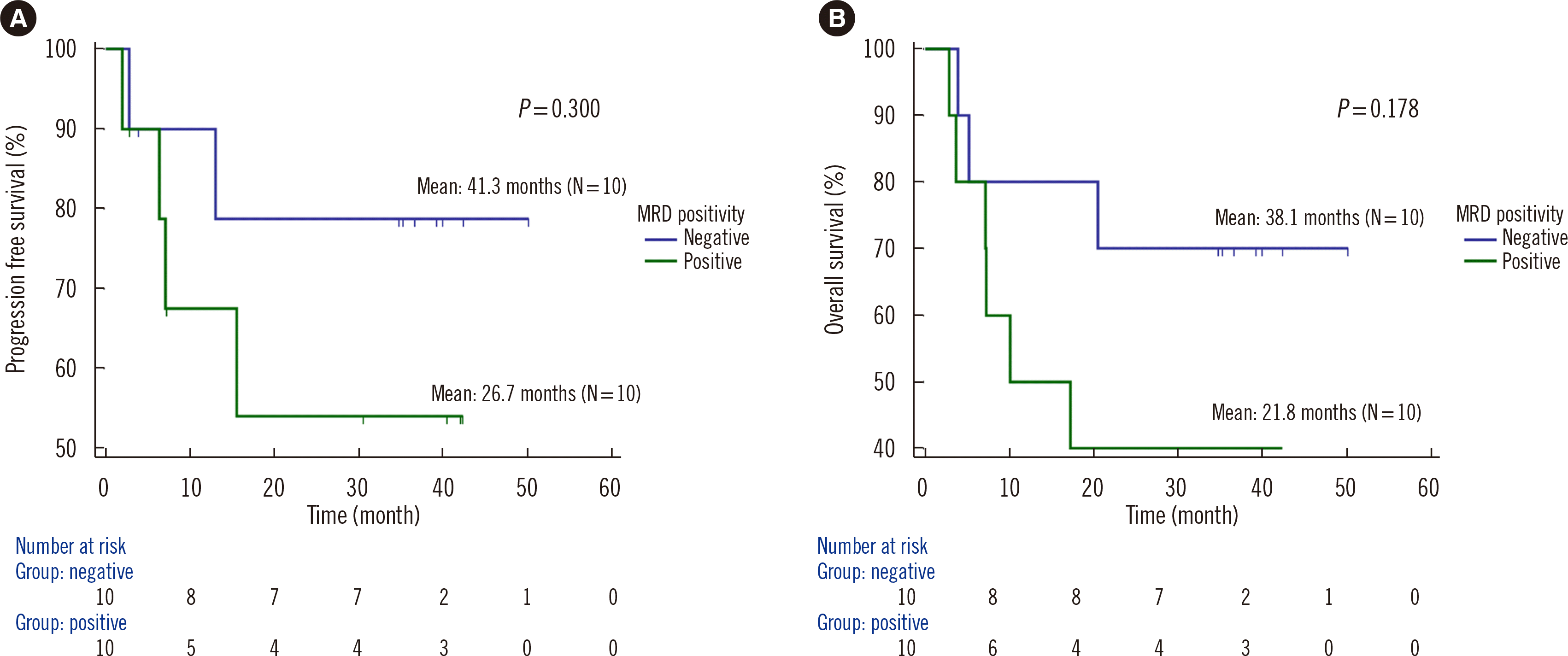
Kaplan–Meier curves for OS and PFS according to MRD positivity in the M1 sample are depicted in Fig. 5. Twenty patients, excluding three lost to follow-up, were evaluated. The mean follow-up period was 945.1 days. All M1 samples with only DNMT3A, TET2, or ASXL1 (DTA) residual mutations were considered MRD-negative as DTA mutations are not associated with the incidence of relapse at any VAF cutoff value [18]. MRD positivity in the M1 sample tended to be associated with poor outcomes in AML patients. The median PFS was 26.7 and 41.3 months for MRD-positive and -negative patients, respectively (P=0.300). The median OS was 21.8 and 38.1 months for MRD-positive and -negative patients, respectively (P=0.178).
DISCUSSION
NGS-based MRD tests have multiple potential applications in the clinical laboratory. AML typically comprises a founding clone and one or more subclones that contain the founding clone mutations plus additional mutations [1, 7]. Key MRD test design challenges revolve around compromises between the breadth and depth of sequencing coverage [19]. Although a broad NGS panel allows monitoring of all clonal mutations in nearly all AML patients, it is limited by a low coverage depth in a wide target space [20], while a high coverage depth is needed for a highly sensitive MRD assessment. Here, we developed a small, cost-effective, and easily adoptable NGS-MRD panel that can be applied to approximately 78.0% of AML patients with somatic mutations identified at diagnosis. The NGS-MRD panel includes JAK2, MPL, and CALR, which are representative myeloproliferative neoplasm (MPN) drivers. Although the frequencies of these genes are low in de novo AML, they can help accurate MPN diagnosis in patients with low-VAF mutations, reveal comprehensive molecular profiles and clonal dynamics associated with disease evolution in high-risk MPN, and track MRD of secondary AML evolving from MPN [21–23]. Our panel can also be applied to some ALL patients, although patient coverage is expected to be low. Further efforts to increase the sensitivity of CNA monitoring with NGS will be needed for MRD monitoring of ALL patients because the prevalence of CNAs (e.g., IKZF1, CDKN2A, CDKN2B, RB1, PAX5, and ETV6) in these patients is high [24, 25].
A key requirement for NGS-based AML MRD tests is high sensitivity or sequencing below the error rate of the sequencing platform, which requires additional steps to improve the error rate and reduce false-positive results [4, 26]. We used the PiSeq computational algorithm to refine the accuracy of molecular barcoding by calculating genome positions of mapped reads to reduce sequencing error rates [15]. Validation using reference material indicated that the MRD panel test was sensitive at a mutation frequency of 0.25% for SNVs. In addition, the VAF obtained with the NGS-MRD panel showed excellent linearity and accuracy compared with that obtained by ddPCR. The ultrasensitive NGS-MRD method can predict patient relapse risk and survival [26–28].
In addition to the design challenges for clinical NGS-based MRD tests, there are interpretive challenges. A major potential pitfall of NGS panel-based MRD interpretation is the persistence of clonal hematopoiesis of indeterminate potential (CHIP) mutations, which include the most commonly mutated candidate driver genes in CHIP, the so-called “DTA mutations” after chemotherapy [18]. Recently, the ELN has recommended not to include DTA variants in NGS-MRD panels for MRD monitoring [29]. In the three patients with only persistent DTA mutations, we found no correlation between mutation status and disease relapse, although the variant frequency ranged up to 21.32%. However, in patients with recurrence, the DTA mutation fraction tended to increase, as did that of accompanied gene mutations. In addition, the fraction of ASXL1 mutations in samples collected at two months was increased in patient #15, who died due to transplant failure. DTA or CHIP mutations tend to be associated with relapse or delayed recovery in AML patients [30, 31]. Therefore, we included the DTA genes in the MRD-NGS panel, but the results should be interpreted with caution. In addition, clinicians can consider additional chemotherapy when the fractions of DTA mutations increase, and more clinical evidence is needed for their prognostic impact.
Although MRD positivity in M1 samples tended to be associated with inferior OS and PFS, the association was not significant. Patients with undetectable mutations have better clinical outcomes than patients with detectable residual mutations [27, 32]. Performing NGS-MRD before HSCT has clinical utility as a negative prognostic indicator [26, 33]. Future studies with larger numbers of patients may clarify the association between MRD positivity and poor patient outcomes.
In two patients with paired samples at diagnosis and relapse (patients #3 and #6), WT1 and NRAS mutations, respectively, emerged in relapsed samples. WT1 mutation is associated with an adverse outcome in AML [34, 35]. In the case of NRAS mutation, the association with prognosis of AML has yet to be clarified [36, 37].
Our study had several limitations. First, the sample size was relatively small. Second, the panel genes were selected based on data from a single institution. Therefore, our NGS-MRD panel does not include some genes suggested in recently updated guidelines, such as the ELN 2021 update on AML MRD (DDX41, EZH2, PTPN11, RAD21, and SRSF2) [29] and the WHO/International Consensus Classification 2022 update classification on AML with myelodysplasia-related gene mutations (EZH2, SRSF2, and ZRSR2) [38, 39]. In the 346 AML patients who underwent baseline NGS at our institute, the detection frequency of the above six genes (DDX41, EZH2, PTPN11, RAD21, SRSF2, and ZRSR2) was 39/346 (11.3%). Most of these patients (31/39, 79.5%) had variants in other genes included in our panel in addition to the six genes; therefore, these patients can be monitored using our panel.
We designed the NGS-MRD panel targeting the entire coding regions of all target genes. However, to maintain sequencing output and increase patient coverage, it may be more efficient and reasonable to include hotspot regions for genes with mutation hotspots (e.g., IDH1/2, JAK2, CALR, MPL, KIT, SF3B1, NRAS, KRAS, FLT3, NPM1, and U2AF1) and to increase the number of other clinically essential genes. Regarding panel contents, each laboratory should review accumulating evidence and update target regions in an ongoing manner [40]. Third, we included only several recurrent SNVs in the analytical performance validation using the reference material, and no other types of variants, such as large insertions (e.g., FLT3-ITD) or variants within homopolymer regions (e.g., ASXL1 c.1934dup), which show reduced detection sensitivity using NGS [41, 42]. Among 19 CR samples derived from eight patients with markers other than FLT3-ITD, five samples tested FLT3-ITD negative and other gene mutations except for DTA tested positive using the NGS-MRD panel. The results for FLT3-ITD and other mutations in the remaining samples were consistent (one positive and 13 negatives). These results suggest that detecting FLT3-ITD by NGS-MRD might be less sensitive than detecting SNVs or small insertions/deletions. Although none of the patients had the ASXL1 c.1934dup mutation at diagnosis, variant calls (average 0.53%, range 0.15%–1.47%) at this location were observed in nearly all CR samples tested (40/48, 83.3%). Therefore, the lower detection limit of the ASXL1 c.1934dup mutation was expected to be substantially higher than the calculated value of 0.25% using the reference material containing several SNVs.
In conclusion, we designed a highly sensitive and accurate NGS-MRD panel to monitor AML patients that can be readily applied in clinical practice. This NGS test can be used in most AML patients, including patients without gene rearrangement. Using the NGS-MRD panel, clones emerging during relapse can be detected early, improving prognoses for AML patients.
 XML Download
XML Download