

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The gut microbiota can be viewed as hidden metabolic organ with an great impact on host metabolism, physiology, nutrition and immune function [1]. At birth, gastrointestinal tract (GIT) of newborn infant is considered as sterile, however, the development of microbiota in the GIT begins when the newly born infant comes in contact with the microorganisms from mother and surrounding environment after birth [2,3]. It has been reported that the rapid colonization of gut begins in the first few days which advances to stabilization in the following months [4,5]. The factors which influence the development of gut microbial community are categorized as: extrinsic factors (such as geographic region, maternal and surrounding bacteria, mode of delivery, hygiene measures, and feeding habits, and drug therapies) and intrinsic factors (such as the neonatal genetics, bacterial mucosal receptors, and interactions, intestinal pH and secretions, peristalsis, and immune response) [5,6].
A review on previously documented literature reveals that rapid and less laborious molecular fingerprinting method such as denaturing gradient gel electrophoresis (DGGE) are widely used to study the dynamics and shifts in gut microbiota based on single nucleotide base separation within the 16S rRNA gene [7,8]. As per the United Nations Children’s Fund (UNICEF) record, India accounts for approximately 20% of the world’s total birth with an infant mortality rate of 5.3% [9]. Despite of such high birth rates and mortality in Indian population, there are no studies on profiling of infant gut microbiota from Indian origin which can provide a cause of these mortalities.
There are only 2 studies reported to analyze the pattern of colonization of gut microbiota in Indian infants [10,11]. Kabeerdoss et al. [10] used qPCR against targeted bacterial population (lactobacilli, bifidobacteria, Clostridia, Bacteroides-Prevotella) in pooled samples of 83 infants with different feeding regimes (breast feeding or formula feeding) highlighting the effect of mode of delivery on microbial succession for initial 6 months. Another study by Pandey et al. [11] reported the effect of mode of delivery on the infants gut microbiota, with only single sample collection i.e. on 7th day after birth. The authors reported the dominance of Acinetobacter sp., Bifidobacterium sp. and Staphylococcus sp. among vaginal delivered infant and Citrobacter sp., Escherichia coli and Clostridium difficile among caesarean section derived infants.
Therefore, there is an urgent need to understand the development of gut microbiota especially from the Indian geographical region with diverse dietary patterns to gain better understanding of colonization patterns of microbial groups in infant gut. Keeping these points in mind the present study aimed to use PCR-DGGE based sequence analysis to compare the diversity and dynamics of the lactic acid producing bacteria (LAB) and bifidobacteria during the initial stages of development of gut microbiome.
METHODS
1. Ethics Statement
All study procedures such as infant stool sample collection, storage and analysis were approved by Institutional Ethical and Biosafety Committee (IEC No. 22/2015). Informed consent was obtained from the parents of the infant in this study.
2. Subjects and Preparation of Samples
Fresh stool sample from 10 full-term, breast fed and vaginally delivered neonates were used in this work. A total of 4 samples were collected in sterile vials weekly from each infant per month resulting in total 40 samples per month. All the 40 samples collected per month were pooled before subjecting for DNA extraction. These samples were stored at –20°C and processed within 3 hours for DNA extraction (Supplementary Table 1).
3. DNA Extraction and PCR
DNA was isolated from 500 mg (wet weight) of stool sample using standard methodology with slight modifications [11]. The total bacterial population as well as specific LAB and bifidobacterial group was amplified using specific primer sets with specific temperature–time programs (Table 1) [12-14]. Before amplification, the DNA was quantified and diluted upto concentration of 100 ng/μL. The 16S rRNA genes total bacterial population was amplified by PCR using the forward primer PRBA338f with GC clamp of 40 bp and the reverse primer P518r. To amplify the 16S rRNA genes of the LAB and bifidobacterial group, a nested PCR approach was used. In the first PCR amplification, group specific primer sets were used whereas in the 2nd PCR amplification, primers PRBA338fGC and P518r were used.
4. DGGE and Sequencing
DGGE was conducted using the DCode™ Universal Mutation Detection system (Bio-Rad, Hercules, CA, USA). PCR products were loaded onto 6% (w/v) polyacrylamide gels using 1× TAE (10 mM acetate, 0.5 mM EDTA, 20 mM Tris, pH 7.4) [15]. For separation of amplified DNA fragments, denaturing gradients ranging from 20% to 45% was used in polyacrylamide gels. The electrophoresis was run at 75 V for 16 hours at 60°C. Staining of gel was done with ethidium bromide solution (0.005%) and image was captured using Alphaimager® EP gel imaging system version 1.3.0.7 (Alpha Innotech, San Leandro, CA, USA). The predominant DGGE bands were excised and eluted in 100 μL sterile deionized water and were kept overnight at 4°C. The eluted DNA was once again confirmed using PCR technique, using primers without GC clamp and sequenced thereafter (1st base sequencing Services, Malaysia). The 16S rRNA gene sequences were analyzed using BLAST search. The sequences were further analyzed using Cluster Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). The gene sequences have been submitted to GenBank database (Table 2).
5. Data Analysis
The DGGE patterns were analyzed using Quantity one software version 4.6.7 (Bio-Rad). The dendrogram of the DGGE bands were generated on the base of unweighted pair-group method average (UPGMA). The richness (S) of the bacterial community was calculated from the number of bands in each lane and further diversity of the microbial community was calculated by the Shannon index of general diversity “H” as H= –Σ(ni/N)log(ni/N), where ni is the intensity of the band and N is the sum of all bands intensity [16]. H was calculated using peak intensity from the different bacterial groups (16S rRNA bands). Furthermore, the richness and evenness (E) of the microbial community were also calculated to measure the number of the species present and to measure the uniform abundance of the different species, respectively. The evenness was calculated as E=H/lnS [17]. The similarity between microbiota compositions at different months was determined by Sorenson’s pair wise similarity coefficient (Cs) [18]. Cs values were determined by the following equation: Cs=ij/(T)×100 where i is the total number of lanes to be compared, j is the number of common DGGE band, T is the total number of DGGE band in all lanes to be compared. The 2 completely identical profiles give 100% similarity whereas completely different give 0% similarity. Comparison of results was done by ANOVA and Tukey’s multiple comparison test (P<0.05) by SPSS statistics 17.0 (SPSS Inc., Chicago, IL, USA).
RESULTS
The present study was conducted to analyze the diversity and establishment of LAB and bifidobacterial communities in healthy full-term breast-fed infants using PCR-DGGE based sequence analysis.
The LAB representing the major dominant bands in DGGE gel were identified by sequence analysis of 16S rRNA gene. Comparative analysis of the sequences, within the National Center for Biotechnology Information (NCBI) database revealed high similarity (≥85%) with LAB. The dominant LAB species, during first 4 months indicated the presence of different Enterococcus sp., Streptococcus sp. and Lactobacillus sp. A detailed overview on the sequence analysis of LAB, observed in the present study is presented in Table 2. Perusal of phylogenetic analysis of the sequences revealed that the LAB being reported in the present study, have previously known to be associated with fecal sample, mother milk and vagina, with the notable exception of species such as Enterococcus sulfureus and E. hermanniensis [19].
Comparative analysis of the 16S rRNA sequences of bifidobacteria, within the NCBI database revealed high similarity (≥95%) (Table 2). Looking at the profile of the Genus Bifidobacterium, Bifidobacterium breve was observed as dominant species during all the 4 months of study. The other closest match for bifidobacteria were identified as B. animalis subsp. lactis, B. choerinum and B. pseudolongum.
Based on the analysis of DGGE fingerprints, biodiversity indices (Shannon diversity index, richness and evenness) were measured which gave quantitative measure of varying diversity of LAB and bifidobacterial species (Table 3). Comparison of richness index (S) i.e. number of LAB and bifidobacteria, revealed higher number of species of LAB as compared to bifidobacteria. However, during the 2nd month, the richness of LAB significantly decreased (P<0.05), with no changes in bifidobacterial group (P>0.05). During the 3rd month, the richness and diversity of both LAB and bifidobacteria remained similar (P>0.05) with a significant increase (P<0.05) in richness of both LAB and bifidobacterial group in 4th month. The Shannon diversity index (H) for both the target bacterial genera followed the similar pattern with highest value of 1.318 for LAB and 1.290 for bifidobacteria in the 4th month (Table 3).
DISCUSSION
Recently documented literature sheds light on the importance of studying the diversity of gut microbiota in human infants [20,21], however, limited information is available on the variability of the gut microbiota during the initial stage of growth (from neonate to infant), more specifically in Indian context, more specifically the north-western Himalayan region. Therefore, the present study was conceptualized to understand the dynamics of initial colonization of gut microbiota using DGGE. For the present study, the fresh stool samples were collected from healthy infants who were, full term, vaginally delivered and breast fed. As the breast fed infants are reported to less prone to allergies and other gut infections [22], therefore, these infants are considered as standard for qualitative and quantitative assessment of healthy gut microbiota. The importance of mode of delivery of neonates was taken as criteria for the selection of such subjects for evaluating the dynamics of LAB and bifidobacteria in the present study [23].
The LAB includes a large number of bacterial genera among which the best known are lactobacilli, lactococci, enterococci, streptococci, Leuconostoc, and pediococci. We have observed the dominance of Enterococcus sp., Streptococcus sp. and Lactobacillus sp. as major LAB members in the infant faeces. The Enterococcus sp. identified in present study gave maximum similarity to E. sulfureus, E. gallinarum, E. faecium and E. hermanniensis which have been previously reported from plant-based food products [24], breast milk [25] and infant’s and healthy stool sample [26,27] and GIT of animals [28]. Among Lactobacillus species, the predominant lactobacilli indicated closest homology to L. ruminis, L. salivarius and L. hayakitensis. L. ruminis and L. salivarius have been reported before in breast milk and stool samples of vaginally delivered infants [29,30]. Among the members of Streptococcaceae, 3 species viz. S. salivarius, S. vestibularis and S. thermophilus were observed. Previous reported literature indicates the presence of S. salivarius and S. vestibularis in infant stool samples [22,27] whereas Pandey et al. [11] reported the presence of S. salivarius in fecal sample of healthy adults. The presence of all these 3 species in breast milk has also been reported by several researchers [22,24,31]. Among bifidobacteria, B. breve has been reported as dominant bifidobacteria in both breast milk and infant faeces [22,25,30]. No effect on mode of delivery was observed on the dominance of B. breve [29]. Kharchenko et al. [32] isolated B. animalis subsp. lactis using culture dependent technique from faeces of new born infant. Similarly, B. pseudolongum was isolated by Vazquez-Gutierrez et al. [33] using enrichment media from infant stool sample. Of the special interest is the association of B. choerinum in infant’s gut since it has not been reported to be associated with human gut, however, there are reports of its presence in animal faeces [34].
The microbial diversity of lactic acid producing bacteria and bifidobacteria in the present study was reported in terms of richness, eveness and Shannon diversity. The results revealed a highly variable and dynamic pattern of LAB and Bifidobacterium species within the first 4 months of development of gut microbiota. A higher richness index (S) was observed for LAB and bifidobacterial group during the 1st month, which decreased in the 2nd month which can be attributed to fact that the members of LAB are dominant in healthy human vagina [35] and this group is assumed to be present in vaginally delivered newborns [36]. Further decrease in LAB during 2nd month might be associated with dominance of bifidobacteria or other microbial groups such as Bacterioides-Prevotella and Clostridia. Later on, higher diversity of LAB and Bifidobacterium species was observed which might be due to only breast feeding as studies have indicated the presence of LAB and Bifidobacterium species in breast milk [30]. The factors such as full term neonates, mode of delivery and breast feeding of infants are known to influence the gut microbial diversity of infants [23], however, in the present study, the influence of these factors was mitigated as the subjects chosen for the present study shared similar characteristics.
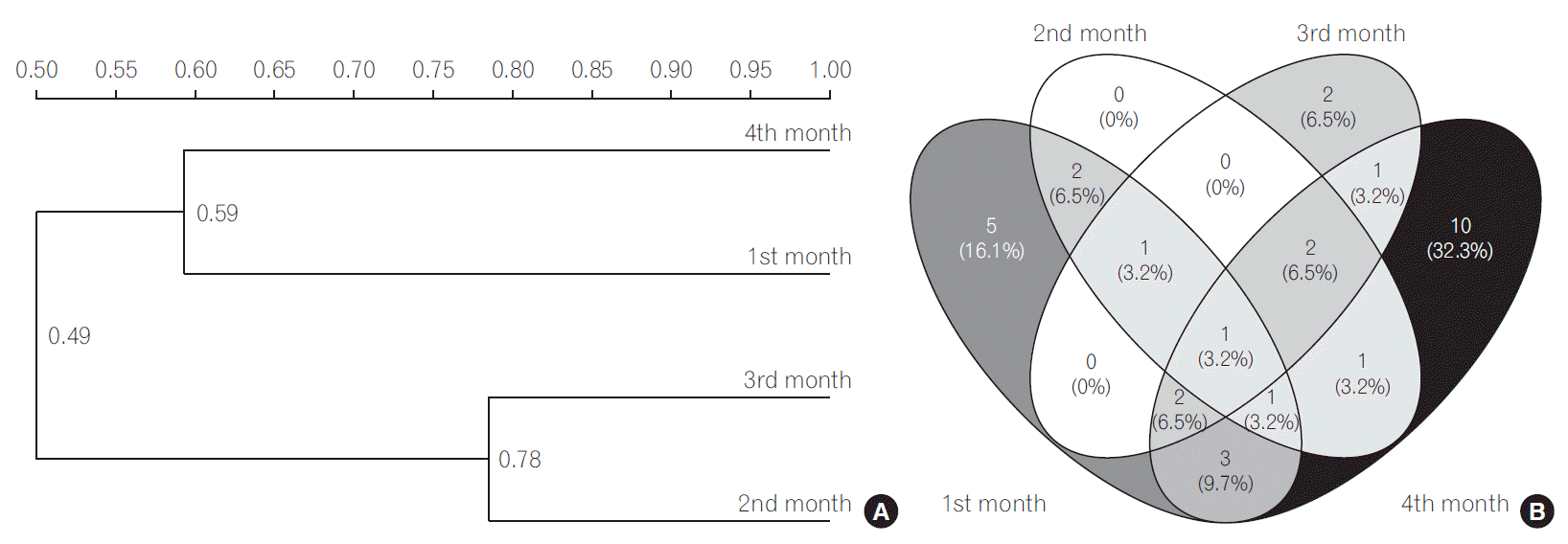
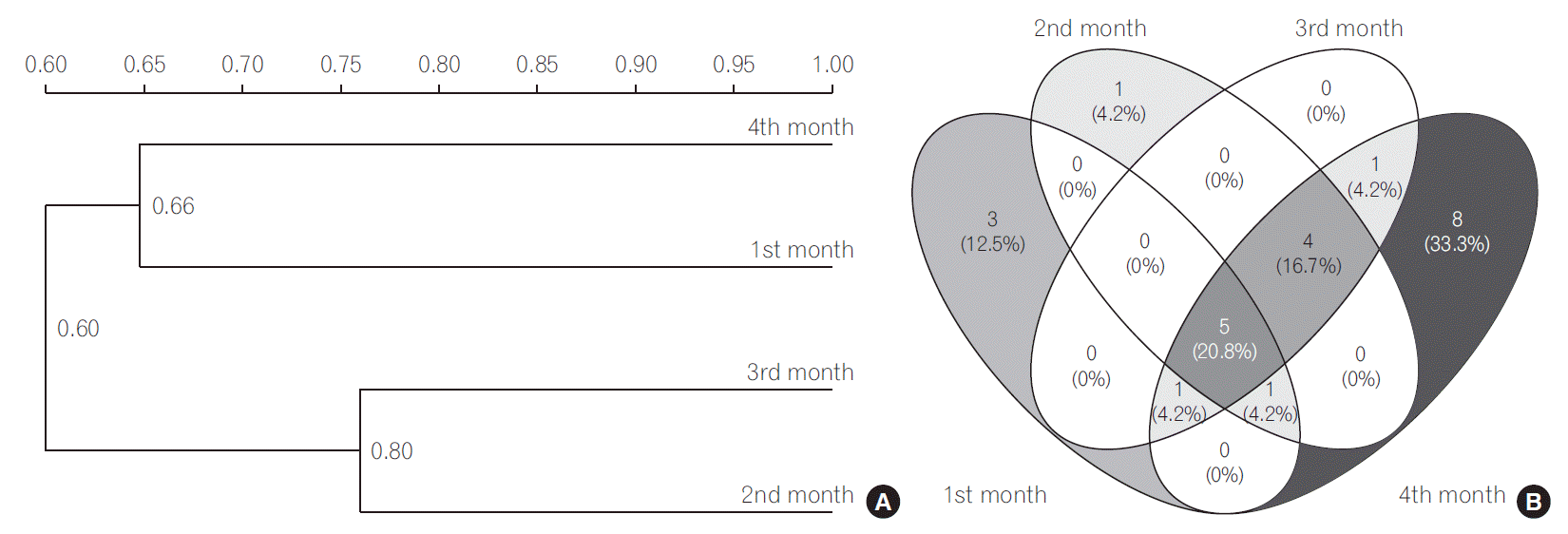
The UPGMA cluster analysis to obtain similarity between LAB and bifidobacteria was conducted. The cluster analysis for LAB indicated high similarity between 3rd and 4th months since they were clustered together (Fig. 1A). The Sorenson’s pair wise similarity coefficient (Cs) revealed a coefficient of 52.17% for 1st and 2nd month, with an increase to 70.58% in 2nd and 3rd months which further decreased to 53.33% for 3rd and 4th months presented as Venn’s diagram (http://bioinfogp.cnb.csic.es/tools/venny/index.html) (Fig. 1B). The Venn’s diagrams were prepared taking into account the number of similar identified species observed in all the 4 months and their relative abundance. The similarity of LAB in different months indicated that 32% of LAB species among all LAB were exclusively determined in the 4th month indicating a stabilized colonization. The cluster analysis of bifidobacterial group based on DGGE profiles (Fig. 2A) revealed clustering of 2nd and 3rd months. The findings indicate that Cs for both LAB and bifidobacteria followed similar trends, with maximum similarities in 2nd and 3rd months (81.81%) and new species in 4th month (Fig. 2B). A gradual increase in the diversity of bifidobacteria with 33% bifidobacterial species exclusively determined in the 4th month was observed indicating a stabilized bifidobacterial population. The study indicated a higher diversity of LAB and bifidobacteria in the 4th month which might be due to consumption of breast milk as feed as the breast milk has been found to be significant source of these microbial groups. Recent documented literature reports the isolation of L. casei, L. gasseri, L. gastricus, L. fermentum, L. plantarum, L. reuteri, L. salivarius, L. vaginalis, B. breve, and B. longum and other members of Staphylococcus, Enterobacteriaceae from human milk [30,37]. Our results are in concomitance with the reports cited above, as is evident from the dominant LAB and bifidobacteria reported in the present study. The breast milk is also reported to contain prebiotic factors such as human milk oligosaccharides (HMOs), antimicrobial proteins (lactoferrin, lysozyme and immunolglobulins) which define the shape of the microbiota in the gut. HMOs are reported to enhance the growth bifidobacterial population. Several in vitro studies have shown that Bifidobacterium sp., such as B. longum subsp. infantis, B. bifidum and B. breve are able to consume HMOs [23,38]; thereby increasing their diversity in gut. Findings in our study also indicate dominance of the above mentioned microbial species indicating a possible role of breast-milk in shaping the gut microbiota.
Thus, based on the findings of this small cohort investigation, we conclude that the gut microbial diversity of LAB and bifidobacteria during the initial phases of development is highly dynamic. A more detailed comparative analysis of gut microbiota with a higher number of subjects and different feeding habits is essential to highlight the contribution of breast milk in development of LAB and bifidobacterial communities. Additionally, an in-depth sequencing including maternal factors needs to be investigated to determine the linkage of transitions in infant gut microbiome to mother phylotypes.
 XML Download
XML Download