

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Trillions of microorganisms colonize various regions of the human body, including the skin, mucosal surfaces, genitals, and gastrointestinal (GI) tract, outnumbering the cell numbers of their human hosts by 10 times.1 Immediately after delivery, newborn infants are exposed to a wide variety of microbes, and their gut microbiota is usually initially dominated by bifidobacteria, ultimately developing into a more diverse characteristic adult pattern.2
3 The composition and function of the gut microbiota rapidly develop to form a complex network that depends on the individual genome, nutrition, antibiotic use, and lifestyle, with variations among ethnicities and countries.4
5
6 The gut microbiota is associated with the regulation of metabolic pathways such as host–microbiota metabolism, signaling, and immune responses that connect the gut, liver, muscle, and brain.7 In addition, dysbiosis of microbial communities in both adults and infants has been shown to be linked to several functional gut disorders including obesity, IBD, type 1 diabetes, autism, necrotizing enterocolitis, and gastric and colon cancers.8
9
10
11
12
13 Several lines of evidence have shown that an abnormal increase or decrease in certain bacteria is associated with health and disease in humans.14
15
16
17
18 Based on these reports, 4 putative genera, Bifidobacterium, Lactobacillus, Bacteroides, and Clostridium, were chosen for analysis. As the gut microbiota plays an essential role in influencing the health of the host, it is important to first understand what constitutes a “healthy” gut microbiome. However, the majority of studies in the literature focusing on the characterization and assembly of the gut microbiota in healthy people have been limited to Western populations, with no extensive investigation of the microbial communities of the Korean population conducted to date.
Therefore, in the present study, we have focused on describing the large-scale profiles that constitute a healthy gut microbiota in Koreans to provide a detailed reference. Previous studies have indicated that the gut microbiota community differs before and after the first year of life.3 Furthermore, the gut microbial composition during infancy can have long-lasting effects on an individual's future health.19
20
21 The primary aim of the present study was to characterize the gut microbiome composition of healthy Koreans, using cultivation-independent molecular techniques. Fecal samples are often used to study the intestinal microflora because they are easily collected noninvasively. Previous studies have evaluated standard microbial cultures using selective media, but these methods lack reproducibility and most bacterial species cannot be cultured. However, progress in bacterial taxonomy based on molecular methods, especially the comparison of the 16S ribosomal RNA (16S rRNA) gene, has made culture-independent investigation the standard approach for identifying and phylogenetically classifying microbial communities.22
23
Although PCR amplification of the 16S rRNA gene in combination with pyrosequencing appears to be a useful approach for studying the microbial composition in an ecological environment such as the human gut, it is limited to providing only an estimation of the relative abundance without information on absolute bacterial density.24 In contrast, quantitative real-time PCR (qPCR) has the advantage of providing more accurate and quantitative information; however, it cannot provide any information on the relative abundance of the total bacterial composition.
Using this approach, we analyzed the gut microbiota of the 2 age groups with respect to the representative genera Lactobacillus, Bifidobacteria, Bacteroides, and Clostridium. The profiles established herein will provide a critical baseline for future studies investigating the Korean gut microbiome composition between infants and adults.
METHODS
1. Subjects
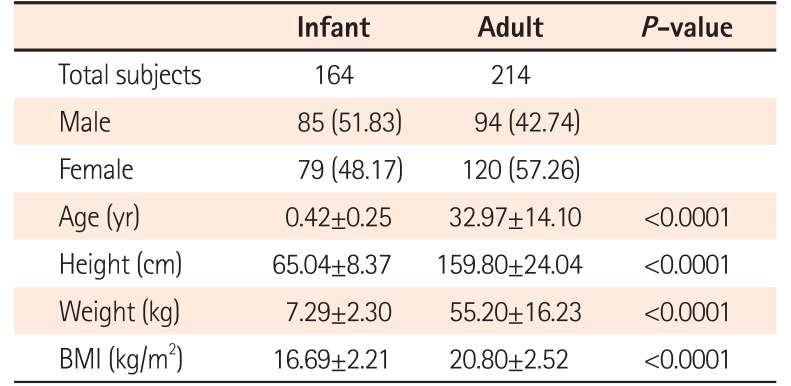
Fecal samples were collected from and donated by 378 healthy Korean individuals (164 infants and 214 adults) recruited at the Samsung Medical Center (Seoul, Korea). Clinical characteristics of the participants are shown in Table 1. The mean age of infants was 0.42±0.25 years, including 85 males and 79 females. The mean age of adults was 32.97±14.10 years, including 94 males and 120 females. Subjects who were overweight or had GI symptoms, a history of GI surgery, asthma, cancer, CD, IBS, IBD, diabetes, and neurological diseases were excluded. In addition, subjects with a history of antibiotic use or intentional probiotic intake were excluded in this study. Written informed consent was obtained from all subjects according to the institutional guidelines, and the protocol was approved by the Institutional Review Board of the participating hospitals (IRB number: 2013-12-061). All methods were performed in accordance with the relevant guidelines and regulations.
2. Sample Collection and Storage
Test kits containing a sterile plastic tube (Sarstedt, Nümbrecht, Germany) with a screw cap to collect the stool samples were provided to the participants. The samples were collected by the participants themselves and immediately stored in home freezers until delivery to the experimental laboratory. The samples were stored at −80℃ until DNA was extracted.
3. DNA Extraction from Fecal Samples
Approximately 200 mg of each stool sample was fully homogenized with 0.1-mm diameter glass beads (Biospec, Orlando, FL, USA). Fecal DNA was subsequently extracted using commercial QIAamp stool DNA extraction kits (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. The concentration and quality of the extracted genomic DNA was determined by agarose gel electrophoresis and spectrophotometric analysis (NanoDrop Technologies, Wilmington, DE, USA). All extracted DNA samples were stored at −20℃ until use in further experiments.
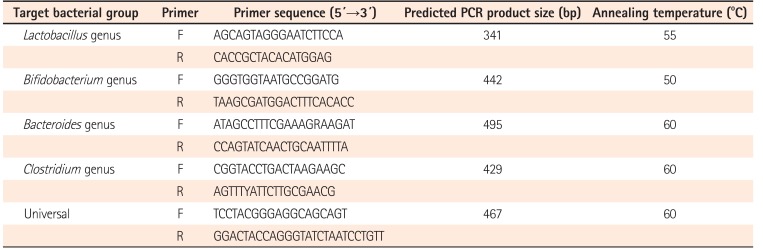
4. Target Bacteria Analysis Using qPCR
To quantify the total bacteria and the bacteria of interest in the stool samples, qPCR was carried out using the ABI SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) with primers amplifying the genes encoding 16S rRNA from specific bacterial groups (Table 2). qPCR for the 4 major genera representative of the gut microbiota (Lactobacillus, Bifidobacterium, Bacteroides, and Clostridium) was performed as described previously25
26
27 to ensure broad bacterial coverage. Each qPCR was carried out in duplicate 20 µL reactions with 1 pM forward and reverse primers, 10 ng purified fecal DNA, and 1X SYBR qRT-PCR Master Mix (Takara Bio Inc., Shiga, Japan). The qPCR conditions were as follows: 4 minutes at 95℃, followed by 40 cycles of 95℃ for 15 seconds, 15 seconds at the appropriate annealing temperature, and 72℃ for 33 seconds. All qPCR plates included a “no-template” negative control for each primer set. All microbiology analyses of fecal samples were performed blindly, without knowledge of the subjects' clinical data. The concentrations of each bacteria in this study were normalized to 16S rRNA expression, the internal reference, in fecal samples using the comparative threshold cycle (CT) method.28 The concentrations of each genus in both groups were calculated as the quantitative abundance of bacterial ΔCT values:
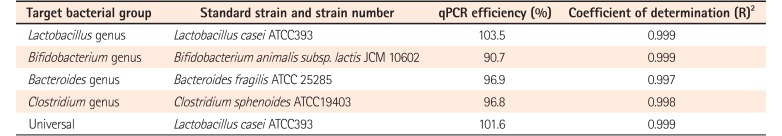
5. Bacterial Strains and Standard Curves for qPCR
To quantify each bacterial group, DNA from the respective reference strain was used for construction of a standard curve. The bacterial strains, efficiency, and coefficient of determination (R2 value) of the standard curve for qPCR are listed in Table 3. Culture conditions used to grow the standard bacterial strains are described in Supplementary Table 1. qPCR was performed as described above with serially diluted bacterial genomic DNA extracted from the reference strains using the QIAamp genomic DNA extraction kit (Qiagen), and the respective copy numbers of 16S rRNA were calculated. Each standard curve was constructed by plotting CT values against bacterial quantity (in 16S rRNA).
6. Statistical Analysis
Clinical characteristics of subjects were expressed as the means±SD. Statistical analysis was performed using Graph-Pad Prism4 (GraphPad Software, San Diego, CA, USA). Abundance in the bacterial species between infants and adults was compared using the Mann-Whitney U test or the Kruskal-Wallis t-test, as appropriate. P<0.05 was regarded as statistically significant.
RESULTS
1. Clinical Characteristics
Fecal samples from 378 healthy Korean subjects were collected for analysis. Table 1 shows the characteristics of the subjects classified into 2 age groups (infants and adults). Statistically significant differences in age, height, weight, and BMI were observed between the 2 groups.
2. The Gut Microbiota Composition
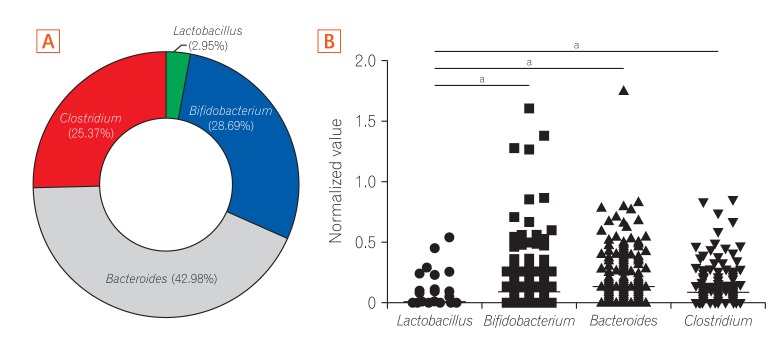
The study participants consisted of 378 healthy Koreans, including 164 infants and 214 adults. In order to analyze the fecal microbiota of this population, we targeted the 4 representative genera of the human gut microbiota to ensure coverage of fecal bacteria composition. Fig. 1 shows the relative abundance of the fecal microbiota groups as determined by qPCR from all subjects enrolled in this study. The most abundant genus overall was Bacteroides, accounting for over 40% of the total microbiota, followed by Bifidobacterium and Clostridium with an average abundance of 28.69% and 25.37%, respectively (Fig. 1A). The proportion of Lactobacillus was below 3% in most of the samples. Lactobacillus was the only group with a significantly lower level (P<0.0001) of normalized copy number by qPCR among the 4 bacteria genera in the overall population sample of healthy Koreans (Fig. 1B).
3. Differences in the Gut Microbiota Composition before and after the First Year of Life
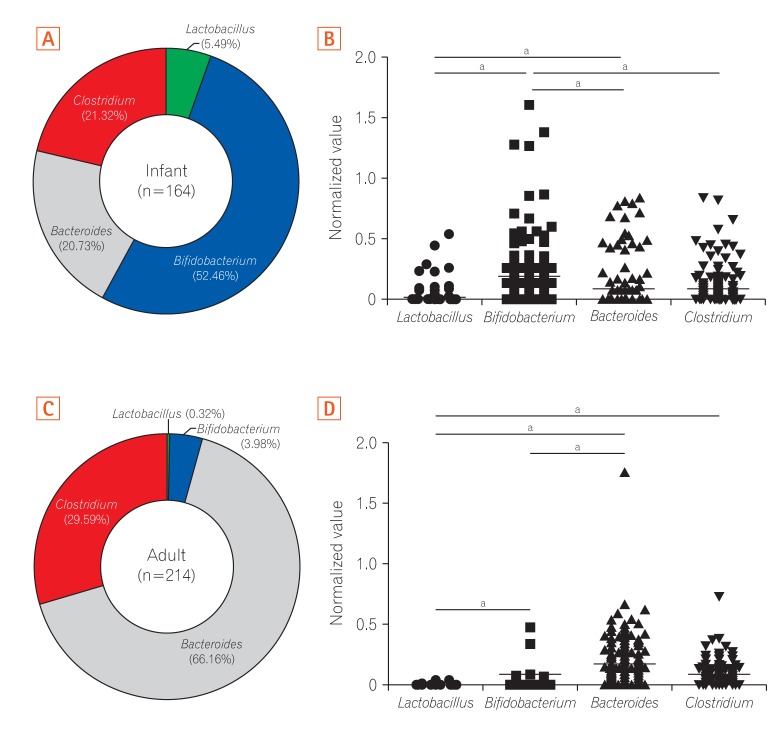
To identify differences in the characteristics of the gut microbiota according to age, we compared the relative composition of each of the 4 genera in infants and adults. According to our predictions, the average proportion of Bifidobacterium was highest (52.46%) in gut microbiota in infants (Fig. 2A), which is in line with previous observations.29
30 The relative abundance of Bifidobacterium was significantly higher than that of the other genera in infants (Fig. 2B). Whereas Bacteroides was the most dominant (66.16%) genus in the gut microbiota of adults (Fig. 2C), which is also supported by previous studies.31
32 When we compared the difference of the gut microbiota composition according to sex, the levels of Lactobacillus and Bacteroides of adult females were significantly higher than those of males (data not shown). There were no significant differences in the composition of the other bacteria between males and females according to relative abundance and normalized values. In adults, the normalized values of Bacteroides and Lactobacillus were significantly higher and lower than those of other genera, respectively (Fig. 2D). Overall, these results indicated that Bifidobacterium is the initial colonizer of the gut microbiota, with a subsequent switch to Bacteroides as the dominant bacteria after the first year of life.
4. Cumulative Analysis of the Gut Microbiota Composition
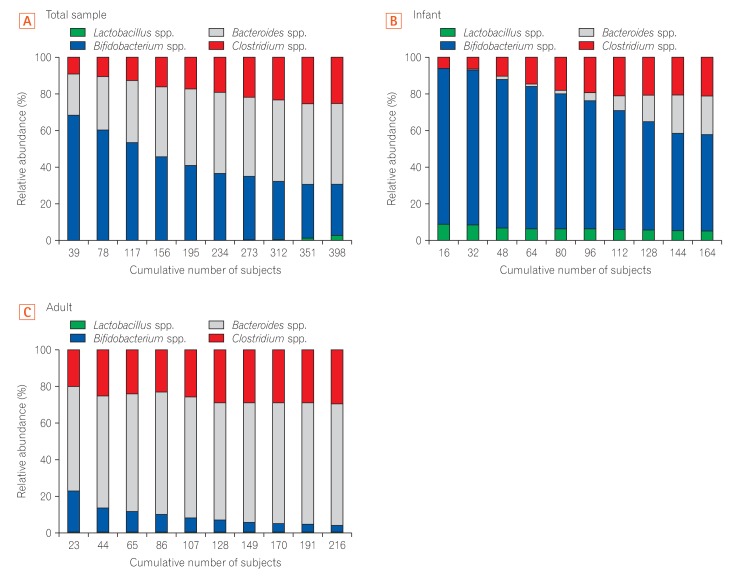
To characterize the composition pattern of the 4 genera according to age in the Korean population, we analyzed the cumulative change in the relative abundance of each genera in the gut of healthy Korean subjects. We classified all Korean subjects on the basis of accumulated values and compared the relative abundance of the gut microbiota for each accumulated point. Although Bifidobacterium was enriched up to a high accumulated value of 30%, Bacteroides gradually became dominant towards the lower accumulated value of the entire sample (Fig. 3A). In contrast, analysis of the gut microbiota composition in infants showed that Bifidobacterium was highly enriched at all accumulated values (Fig. 3B). The proportion of Lactobacillus was lowest in the total sample and in adults. In contrast, Bifidobacterium was low and the proportion of Bacteroides was high at all accumulated values in the gut microbiota community in adults (Fig. 3C). In all cumulative graphs, Clostridium became gradually more enriched towards the lower accumulated value.
DISCUSSION
During adult life, the community composition of the gut microbiota could be influenced by host genetics, health, diet, probiotics, and aging.8
33 Although there have been several reports on investigations of the gut microbiome in Western and in some Asian populations,8
32
34 little is known about the healthy Korean gut microbiota. To identify the gut microbiome composition of the healthy Korean population, in the present study, we analyzed the gut microbiota composition of 378 Koreans using a molecular approach. Indeed, several bacteria that had never been isolated from human feces were detected by PCR in the present study. Previous studies have analyzed the levels of Lactobacillus, Bifidobacterium, Bacteroides, and Clostridium genera that commonly inhabit in the human colon in feces by PCR.14
35
36
According to previous clinical studies, the levels of certain bacteria may vary in health and disease.15
16
17
18
37
38
Lactobacillus is the predominant beneficial bacteria in adults. The level of Lactobacillus is generally lower in patients with IBS than in healthy controls.39
Bifidobacterium have been considered to be the most essential organisms for infants and also constitute one of the most important organisms in the colonic flora of healthy children and adults.3 In particular, the abundance of Bifidobacterium is decreased in patients with some functional GI disorders such as IBS and IBD.15
18 Some Lactobacillus and Bifidobacterium strains are used as probiotics, showing a beneficial effect in the prevention of inflammation in the intestine16 and the ability to eliminate harmful factors impeding the repair of damaged tissue. Hence, probiotics play a key role in aging, regulation of brain gene expression, and inflammation.17
40 Dethlefsen et al.33 found that a small number of genera within the Bacteroidetes and Firmicutes phyla accounted for the majority of gut microbial communities. Bacteroides and Clostridium belong to the phyla Bacteroidetes and Firmicutes, respectively and have been found to be increased in abundance in patients with IBS.37
38 These studies imply that changes in these 4 bacterial genera: Lactobacillus, Bifidobacterium, Bacteroides, and Clostridium are closely related to human health and disease.
Based on this background, we examined the average distributions of Lactobacillus, Bifidobacterium, Bacteroides, and Clostridium in the gut microbiota of healthy Koreans. The most abundant genus in the total samples was Bacteroides, at 42.98%, followed by Bifidobacterium and Clostridium at 28.69% and 25.37%, respectively. Nam et al.41 also reported that Bacteroides was 1 of the 3 most abundant genera in the Korean gut microbiota. Moreover, Arumugam et al.42 found that levels of Bacteroides were relatively high in the fecal metagenomes of individuals from 4 countries, thus classifying Bacteroides as an enterotype.
The diversity of the genus Bifidobacterium in the infant gut microbiota is currently not well understood. In culture-based studies of the gut microbiota associated with aging, Mitsuoka3 showed that Bifidobacteria were the major organisms in the colonic flora of healthy children and adults. Similarly, the fecal samples of the infants in the present study showed a predominance of Bifidobacterium using a cultureindependent system. In agreement with our results, Turroni et al.43 also found a large proportion of Bifidobacterium in the gut microbiota of 11 infants. These studies reinforce the notion that Bifidobacteria are a predominant component of the infant gut microbiota. Bacterial populations within the infant gut are influenced by many factors such as mode of delivery, breastfeeding versus formula feeding, and the use of antibiotics.44
45 Infants tend to have an unstable, less diverse gut microbiota composition than adults. Following the consumption of solid food, the gut microbial community of the infant begins to develop towards the adult pattern, which differs from that of infants. Indeed, in our study, adults typically had a gut community dominated by Bacteroides . Surprisingly, we confirmed that the gut microbiota composition of our relatively small random sample showed a similar distribution of other bacteria including Bacteroides in this population. These findings strongly support the notion that healthy Koreans tend to show a common gut microbiota composition.
Taken together, we have demonstrated that the average relative abundance of 4 genera in the gut microbiota can be analyzed at a large scale in healthy Koreans using a culture-independent method from fecal samples. Although we could not evaluate the effects of dietary patterns and environmental factors on the microbiota patterns identified in the present study, we believe that these factors should be taken into consideration in future studies. As only a limited number of individuals have been subjected to GI microbiota analysis thus far, our data contribute to gaining a comprehensive understanding of the gut microbiota composition of healthy Koreans. These results will be beneficial to future evaluations of the causal relationship between gut microbiota and various diseases, such as IBD, obesity, colon cancer, asthma, and atopy in Korean patients, and should help to establish a reference for the gut microbiota composition in healthy Korean individuals.
 XML Download
XML Download